Abstract
Noroviruses (NVs) are responsible for most cases of human nonbacterial gastroenteritis worldwide. Some parameters for the purification of NV virus-like particles (VLPs) such as ease of production and yield were studied for future development of vaccines and diagnostic tools. In this study, VLPs were produced by the expression of the VP1 and VP2 gene cassette of the Brazilian NV isolate, and two purification methods were compared: cesium chloride (CsCl) gradient centrifugation and ion-exchange chromatography (IEC). IEC produced more and purer VLPs of NV compared to CsCl gradient centrifugation.
Key words:
Norwalk virus; VLP; ion-exchange chromatography
Human noroviruses (HuNVs) are responsible for major outbreaks of nonbacterial gastroenteritis for all ages worldwide. HuNVs cause acute gastroenteritis in schools, kindergartens, homes for the aged, cruise ships and even hospitals. A million cases noroviral infection are verified by laboratory diagnosis each year globally, and 110,000 and 19 million infections were observed last year in Germany and the United States (CDC, 2014), respectively. A large norovirus gastroenteritis outbreak occurred in the Brazilian state of São Paulo in the summer of 2010 (Morillo et al., 2011Morillo SG, Luchs A, Cilli A et al. (2011). Large gastroenteritis outbreak due to Norovirus GII in São Paulo, Brazil, summer 2010. Rev Inst Med Trop 53:119-120.).
The HuNVs are classified into genogroups I (GI), II (GII) and IV (GIV) of Norwalk virus species. GI currently has nine genotypes, GII has 22 and GIV has two(LaRosa et al., 2008La Rosa, Pourshaban M, Iaconelli M et al. (2008) Detection of genogroup IV noroviruses in environmental and clinical samples and partial sequencing through rapid amplification of cDNA ends. Arch Virol 153:2077-2083; Kroneman et al., 2013Kroneman A, Vega E, Vennema H et al. (2013) Proposal for a unified norovirus nomenclature and genotyping. Arch Virol 158:2059-2068.). Among these genotypes, GII.4 is responsible for most outbreaks detected in more than 60-80% of cases of acute gastroenteritis caused by NVs(Lindesmith et al., 2008Lindesmith LC, Donaldson EF, Lobue AD et al. (2008). Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med 5:e31.). In Brazil, the surveillance of acute diarrhea by the Ministry of Health still does not include this agent, so the impact of HuNV on public health in Brazil is being studied for research groups in states such as Rio Grande do Sul and Pará (Fioretti et al., 2011Fioretti JM, Ferreira MSR, Victoria M et al. (2011) Geneticdiversity of noroviruses in Brazil. Mem Inst Oswaldo Cruz 106:942-947.; de Andrade et al., 2014de Andrade Jda S, Rocha MS, Carvalho-Costa FA et al. (2014) Noroviruses associated with outbreaks of acute gastroenteritis in the State of Rio Grande do Sul, Brazil, 2004-2011. J Clin Virol 61:345-352.; Siqueira et al., 2013Siqueira JA, Linhares Ada C, de Carvalho TC et al. (2013) Norovirus infection in children admitted tohospital for acute gastroenteritis in Belém, Pará, Northern Brazil. J Med Virol 85:737-744.).
Virus-like particles (VLPs) prepared by expressing capsid genes using a baculoviral expression system is a useful tool for studying uncultivable viruses such as HuNVs. HuNV VLPs and the native particles have very similar physicochemical and antigenic characteristics (Herbst-Kralovetz et al., 2010Herbst-Kralovetz M, Mason HS, Chen Q (2010) Norwalk virus-like particles as vacines. Expert Rev Vaccines 9:299-307.; Fang et al., 2013Fang H, Tan M, Xia M et al. (2013) Norovirus P Particle Efficiently Elicits Innate, Humoral and Cellular Immunity. PlosOne 8:e63269.). The VLPs have thus been used for immunological studies producing poly-and monoclonal antibodies against HuNV VLPs and also for the search for the cellular receptors of NVs (Herbst-Kralovetz et al., 2010Herbst-Kralovetz M, Mason HS, Chen Q (2010) Norwalk virus-like particles as vacines. Expert Rev Vaccines 9:299-307.; Tan et al., 2011Tan M, Huang P, Xia M et al. (2011) Norovirus P particle, a novel platform for vaccine development and antibody production. J Virol 85:753-764.). Immunological diagnostic tools such as ELISAs are very useful in public health, but Brazil does not yet have a good and abundant source of antibodies for NV diagnostics.
The self-assembling norovirus virus-like particles with capsid protein expression has spurred the development of vaccines, because the particles are assembled similarly to the native viruses and do not cause infection. The intranasal and oral administration of VLPs has been used for inducing mucosal immunity against NV infection. Furthermore, these particles are being used as tools to elucidate the role of immune cells such as T and B lymphocytes in infections caused by this virus. Studies of NV VLP vaccines have focused on a single genogroup, but multivalent vac-cines using different genogroups are more interesting for a broader protection (Sundararajan et al., 2015Sundararajan A, Sangster MY, Frey S et al. (2015) Robust mucosal-homing antibody-secreting B cell responses induced by intramuscular administration of adjuvanted bivalent human norovirus-like particle vaccine. Vaccine 33:568-576.).
The objective of this study was to establish VLPs of NV GII.4 by the expression of the VP1 and VP2 gene cassette for future use as antigens for the development of diagnostic tools. The ease and efficiency of VLP purification were also assessed by comparing ultracentrifugation using cesium chloride and ion-exchange chromatography.
One fecal suspension positive for HuNV was obtained from a seven-year-old child at the laboratory of Virology, Central Public Health Laboratory of the Federal District (LACEN-DF), during a research project approved by the Ethics Committee in Research for protocol Nº SES/DF #93/09 on 11/05/2009. Total RNA was extracted from this sample using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufactures instruction. Complementary DNA (cDNA) was obtained by reverse transcription using Superscript III (Invitrogen) with an oligo-dT primer (Oligo dT50M10: AAG CAG TGT TAT CAA CGC AGA T50). The cDNA fragments of approximately 2.4 kb containing the complete VP1 and VP2 capsid genes and the 3’UTR(untranslated region) were amplified by PCR using NoroII-BamHI forward primer (TTT GGA TCC ATG AAG ATG GCG TCG AAT GAC) and PstI-SacI M10 reverse primer (TAT TCT GCA GAG CTC AAG CAG TGT TAT CAA CGC AGA). The PCR was performed using LongAmp Taq DNA polymerase (New England Biolabs, NEB, Ipswich, MA, USA). The PCR mixture was prepared as follows: 5 μL of 5X LongAmp Taq Reaction Buffer (NEB), 3 μL of 2.5 mM dNTPs, 0.5 μL each of forward and reverse primers (10 mM), 1 μL of cDNA, 1 μL of LongAmp Taq DNA polymerase (2 U) and Milli-Q water (Whatman-GE Healthcare Life Sciences, Piscataway, NJ, USA) to 25 μL. The cycle was set as follows: pre-cycle denaturation step of 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 56 °C for 30 s and extension at 65 °C for 3 min and a final extension at 65 °C for 10 min.
The amplified cDNA containing the VP1 and VP2 genes and the 3’UTR was cloned in the pGEM-T Easy plasmid (Promega, Madson, WI, USA) and subcloned in the baculoviral system plasmid pFASTBac1 (Invitrogen, Carlsbad, CA, USA). The cloning of this bicistronic gene cassette by ribosomal termination-reinitiation was confirmed by sequencing by Macrogen Inc. (Seoul, Korea). The expression of VP1 is sufficient for VLP assembly, but the expression of the VP1-VP2-3’UTR gene cassette is a better strategy for stable and efficient VLP formation (Bertolotti-Ciarlet et al., 2003Bertolotti-Ciarlet A, Crawford SE, Hutson AM et al. (2003). The 3’ end of norwalk virus mRNA contains determinants that regulate the expression and stability of the viral capsid protein VP1: a novel function for the VP2 protein. J Virol 77:11603-11615.; Vongpunsawad et al., 2013Vongpunsawad S, Venkataram Prasad BV, Estes MK (2013) Norwalk Virus Minor Capsid Protein VP2 Associates within the VP1 Shell Domain. J Virol 87:4818-4825.). After constructing the bacmid using the baculoviral Bac-to-Bac system (Invitrogen) according to the manufacturer's instructions, we produced the VP1 and VP2 proteins in the insect cell lines BTI-4-Tn5B1, Sf9 and Sf21.
Preliminary testing indicated that VP1 expression level was higher in the BTI-Tn5B1-4 than the Sf9 and Sf21 cells. VP1 is secreted into the culture medium in the baculoviral expression system, so VLP purification began with the culture medium of the infected cells. The culture medium (105 mL) was centrifuged at 5,000 x g at 4 °C for 10 min to remove the cells, and the supernatant was ultracentrifuged with 20% sucrose cushion at 115,878 x g for 1 h at 4°C. The pellet was resuspended in 0.5X PBS buffer (pH 5.5), and the solution was added to the top of a cesium chloride solution (CsCl 0.39 g/mL) diluted in 0.5X PBS pH 5.5, following the method by Ausar et al., 2006Ausar SF, Foubert TR, Hudson MH et al. (2006) Conformational Stability and Disassembly of Norwalk Virus-like Particles. J Biol Chem 281:19478-19488.. The sample was ultracentrifuged at 114,600 x g for 18 h at 4 °C. The upper, middle and lower fractions were collected and dialyzed three times in 0.5X PBS pH 5.5 with 3 h intervals. The samples were analyzed by SDS-PAGE and Western blotting using a polyclonal antibody against NV (GII.4).
VLP assembly in the purified preparation was confirmed by transmission electron microscopy. A formvarcoated nickel grid (200 mesh) was incubated over 40 μL of the purified VLP solution in PBS for 5 min, and the sample grids were contrasted with 1% uranyl acetate for 5 min, then air dried. The VLPs were observed with a JEM 1011 electron microscope (JEOL, Tokyo, Japan).
The VLPs were also purified by ion-exchange chromatography for comparison. After the 20% sucrose-cushion ultracentrifugation, the pellet was resuspended with sodium phosphate buffer (50 mM, pH 7.0). The VLPs were purified under appropriate conditions by an ÄKTA purifier (GE Life Sciences, Piscataway, NJ, USA) using a HiTrap Q FF column (GE Life Sciences) at a flow rate of 150 cm/h (Koho etal., 2012Koho T, Mäntylä T, Laurinmäki P et al. (2012) Purification of norovirus-like particles (VLPs) by ion exchange chromatography. J Virol Methods 181:6-11.). The bound protein was eluted with stepwise increments in ionic strength to 1 M NaCl, 50 mM sodium phosphate buffer, pH 7.0. The NV VP1 fractions were collected with increasing salt concentrations of 10, 20 and 40%. After purification by both methods (CsCl isopycnic centrifugation and Ion-exchange chromatography), the proteins were quantified by the NanoDrop 3300 Quant-iT Protein Assay (Invitrogen).
No bands were observed from CsCl ultracentrifugation, perhaps due to the low concetration of VLPs. The top, middle and bottom fractions were again ultracentrifuged to precipitate possible VLPs in each fraction. All three fractions were subjected to SDS-PAGE and subsequent Western blotting. The SDS-PAGE produced protein bands of approximately 60 kDa, close to the expected size of 59 kDa (Figure 1A). These proteins, however, were not the target proteins. The Western blotting identified specific signals only for the middle and bottom fractions of the CsCl ultracentrifugation (Figure 1B, lanes 4 and 5) but not for the negative control and the top fraction (Figure 1B). Two specific signals of approximately 59 kDa and 56 kDa were identified, which are the common size so f VP1 produced by the baculoviral expression system (Figure 1B arrow). The smaller protein was the cleaved VP1 lacking the N terminus (Ausar et al., 2006Ausar SF, Foubert TR, Hudson MH et al. (2006) Conformational Stability and Disassembly of Norwalk Virus-like Particles. J Biol Chem 281:19478-19488.). We thus concluded that VP1 is an insoluble protein, which can be separated by ultracentrifuga tion with a 20% sucrose cushion and with a buoyant density heavy enough to remain in the middle and bottom fractions of the CsCl gradient. The dispersed distribution of VLPs in both fractions, however, suggested the failure of gradient formation in the CsCl isopycnic centrifugation under the conditions used.
Analysis of fractions for VP1 proteins by SDS-PAGE and Western blotting prepared by CsCl isopycnic centrifugation. (A) SDS-PAGE gel stained with Coomassie Brilliant Blue and (B) Western blotting using a polyclonal antibody against NV GII.4. Lane 1, PageRuler Prestained Protein Ladder (Thermo Scientific); lane 2, uninfected insect cells (negative control); lane 3, top fraction collected from CsCl centrifugation; lane 4, middle fraction; lane 5, bottom fraction. The arrow in B indicates the specific reaction by Western blotting using the antibody against NV.
The possibility of the presence of VP1 in the cultured cells was also evaluated. The contents of the collected cells were also subjected to SDS-PAGE and Western blotting after boiling for 5 min with Laemmli buffer containing SDS, but almost no VP1 was detected from the infected cells (data not shown).
To determine if the purified VP1 assembled VLPs, we prepared grids with these two fractions. Round particles with protruding structures, which are typical of the VLPs of NV were observed by transmission electron microscopy (Figure 2A). All VLPs had negatively stained central areas. The diameter of the VLPs was estimated at approximately 40 nm, indicating T = 3 icosahedral particles. Natural virus particles and adequate VLPs are T=3 icosahedral capsids containing 180 capsomeres of the VP1 protein, with a diameter of approximately 40 nm (Tresset et al., 2013Tresset G, Decouche V, Bryche JF et al. (2013) Unusual self-assembly properties of Norovirus Newbury2 virus-like particles. Arch Biochem Biophys 537:144-152.), but artificial VLPs produced by VP1 expression also often make T = 1 icosahedral particles of 23 nm. In our study, no T = 1 particles were observed, indicating that the amino acid sequence of the VP1 in this study showed the stable characteristics of VP1 proteins for VLP assembly.
Electron micrographs showing the virus-like particles (VLPs) of NV with diameters of approximately 40 nm by negative staining. VLPs prepared by A) CsCl isopycnic centrifugation and B) ion-exchange chromatography. Scale bar = 100 nm.
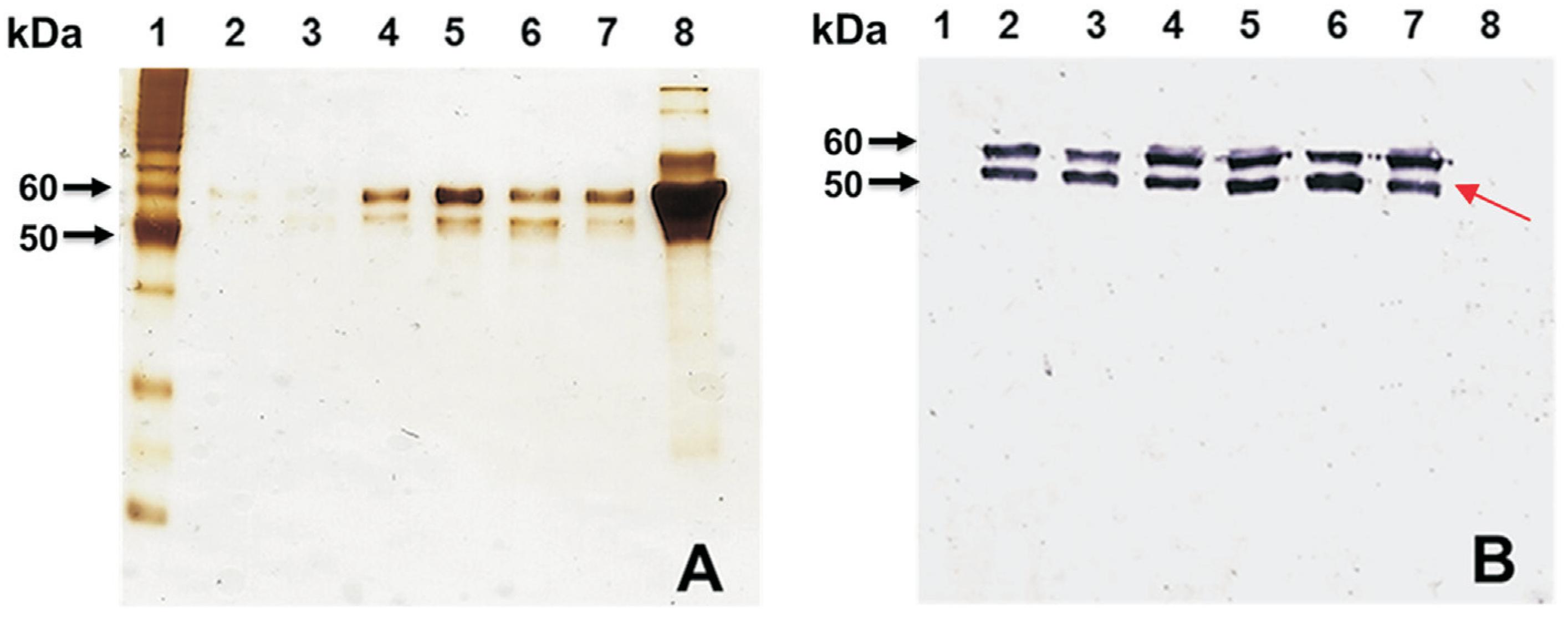
To determine relative VLP yields, we compared ion-exchange chromatography to CsCl centrifugation. The collected fractions were subjected to SDS-PAGE and Western blotting (Figure 3). For visualizing the small amounts of contaminant proteins, the SDS-PAGE gel was silver stained, which is more sensitive than staining with Coomassie Brilliant Blue (Figure 3A). Double protein bands above the 50 kDa indicator protein were observed in the SDS-PAGE (Figure 3A). The identities of these protein bands as VP1 were confirmed by Western blotting (Figure 3B).
Analysis of fractions of VP1 proteins by SDS-PAGE and Western blotting prepared by ion-exchange chromatography. (A) Silver-stained SDS-PAGE gel and (B) Western blotting using a polyclonal antibody against NV GII.4. Lane 1, unstained Novex Sharp Protein Standard ladder (Invitrogen); lanes 2, 3 and 4, fraction 14 of the chromatogram after IEC; lanes 5, 6 and 7, fraction 15 after IEC; lane 8, uninfected insect cell lysate (negative control). Note: the proteins in the uninfected cell lysate identified by silver staining was not reactive in the Western blotting. The arrow in B indicates the specific reaction by Western blotting using the antibody against NV.
The relative strengths of the signals in Figures 1B and 3B indicated that the yield of VP1 was higher by this method than by CsCl ultracentrifugation. The formation of VLPs was also confirmed by electron microscopy (Figure 2B). Abundant VLPs were observed in this preparation and the amount of VLPs obtained by ion-exchange chromatography was greater than that obtained by CsCl centrifugation. The average yield of purified VLPs was 1.50 mg/L by CsCl centrifugation and 9.31 mg/L by ion-exchange chromatography so the yield was 6.2-fold higher for ion-exchange chromatography than for CsCl centrifugation. We also concluded that ion-exchange chromatography produced purer VLPs than the CsCl gradient under our conditions. NV VLP purification by ion-exchange chromatography is recommended over CsCl centrifugation and laboratory-scale VLP purification by CsCl ultracentrifugation often produces sufficient purity and yield, but the procedure requires long ultracentrifugation times. Furthermore, this purification procedure may affect the morphological integrity, functionality and antigenicity of the purified VLPs (Huhti et al., 2010Huhti L, Blazevic V, Nurminen K et al. (2010) A comparison of methods for purification and concentration of norovirus GII-4 capsid virus-like particles. Arch Virol 155:1855-1858.). Ion-exchange chromatography avoids these negative effects thus we concluded that ion-exchange chromatography was the better of the two methods in terms of quality and quantity of VLPs. In addition, purification by ion-exchange chromatography can be advantageous for large-scale VLP production for vaccine development.
This study describes a convenient procedure for the preparation and purification of NV VLP as a first step toward NV surveillance in Brazil. Further development of tools for studying NV is required to improve the epidemiological study of NV in Brazil.
Acknowledgments
We thank the Microscopic Laboratory University of Brasilia and also especially thank Dr. Shinichi Kobayashi (Aichi Prefectural Institute of Public Health, Nagoya, Japan) for kindly providing the polyclonal antibody against NV GII.4 VLPs. “Conselho Nacional de Desenvolvimento Científico e Tecnológico” (CNPq, Brazil) for their financial support of EFN, BMR and TN.
References
- Ausar SF, Foubert TR, Hudson MH et al. (2006) Conformational Stability and Disassembly of Norwalk Virus-like Particles. J Biol Chem 281:19478-19488.
- Bertolotti-Ciarlet A, Crawford SE, Hutson AM et al. (2003). The 3’ end of norwalk virus mRNA contains determinants that regulate the expression and stability of the viral capsid protein VP1: a novel function for the VP2 protein. J Virol 77:11603-11615.
- Centers for Disease Control and Prevention (2014) Burden of norovirus illness and outbreaks. Centers for Disease Control and Prevention, Atlanta.
- de Andrade Jda S, Rocha MS, Carvalho-Costa FA et al. (2014) Noroviruses associated with outbreaks of acute gastroenteritis in the State of Rio Grande do Sul, Brazil, 2004-2011. J Clin Virol 61:345-352.
- Fang H, Tan M, Xia M et al. (2013) Norovirus P Particle Efficiently Elicits Innate, Humoral and Cellular Immunity. PlosOne 8:e63269.
- Fioretti JM, Ferreira MSR, Victoria M et al. (2011) Geneticdiversity of noroviruses in Brazil. Mem Inst Oswaldo Cruz 106:942-947.
- Herbst-Kralovetz M, Mason HS, Chen Q (2010) Norwalk virus-like particles as vacines. Expert Rev Vaccines 9:299-307.
- Huhti L, Blazevic V, Nurminen K et al. (2010) A comparison of methods for purification and concentration of norovirus GII-4 capsid virus-like particles. Arch Virol 155:1855-1858.
- Koho T, Mäntylä T, Laurinmäki P et al. (2012) Purification of norovirus-like particles (VLPs) by ion exchange chromatography. J Virol Methods 181:6-11.
- Kroneman A, Vega E, Vennema H et al. (2013) Proposal for a unified norovirus nomenclature and genotyping. Arch Virol 158:2059-2068.
- La Rosa, Pourshaban M, Iaconelli M et al. (2008) Detection of genogroup IV noroviruses in environmental and clinical samples and partial sequencing through rapid amplification of cDNA ends. Arch Virol 153:2077-2083
- Lindesmith LC, Donaldson EF, Lobue AD et al. (2008). Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med 5:e31.
- Morillo SG, Luchs A, Cilli A et al. (2011). Large gastroenteritis outbreak due to Norovirus GII in São Paulo, Brazil, summer 2010. Rev Inst Med Trop 53:119-120.
- Siqueira JA, Linhares Ada C, de Carvalho TC et al. (2013) Norovirus infection in children admitted tohospital for acute gastroenteritis in Belém, Pará, Northern Brazil. J Med Virol 85:737-744.
- Sundararajan A, Sangster MY, Frey S et al. (2015) Robust mucosal-homing antibody-secreting B cell responses induced by intramuscular administration of adjuvanted bivalent human norovirus-like particle vaccine. Vaccine 33:568-576.
- Tan M, Huang P, Xia M et al. (2011) Norovirus P particle, a novel platform for vaccine development and antibody production. J Virol 85:753-764.
- Tresset G, Decouche V, Bryche JF et al. (2013) Unusual self-assembly properties of Norovirus Newbury2 virus-like particles. Arch Biochem Biophys 537:144-152.
- Vongpunsawad S, Venkataram Prasad BV, Estes MK (2013) Norwalk Virus Minor Capsid Protein VP2 Associates within the VP1 Shell Domain. J Virol 87:4818-4825.
Publication Dates
-
Publication in this collection
09 Oct 2015 -
Date of issue
Oct-Dec 2015
History
-
Received
05 Nov 2014 -
Accepted
12 Apr 2015