caso especial
Deficiência Múltipla Hipotálamo-Hipofisária: Defeito no Gene "Prophet of Pit-1" (Prop-1)
Maria Lúcia C.C. Giannella
Daniel Giannella Neto
Serviço de Endocrinologia e
Metabologia, Hospital das Clínicas,
Faculdade de Medicina da
Universidade de São Paulo,
São Paulo, SP.
Recebido em 02/03/1999
Revisado em 14/07/1999
Aceito em 10/01/2000
O OBJETIVO DESTA APRESENTAÇÃO é a descrição de uma família que apresenta três indivíduos afetados (duas irmãs e um irmão, filhos de pais consangüíneos, primos de primeiro grau) portadores de deficiência combinada de hormônios hipofisários secundária a mutação no gene Prop-1., cuja investigação clínica, laboratorial e radiologica foi complementada pela análise molecular.
Unitermos: Deficiência múltipla hipotálamo-hipofisária; Mutação no gene PROP-1; Deficiência de crescimento; Deficiência de GH.
APRESENTAÇÃO DOS CASOS
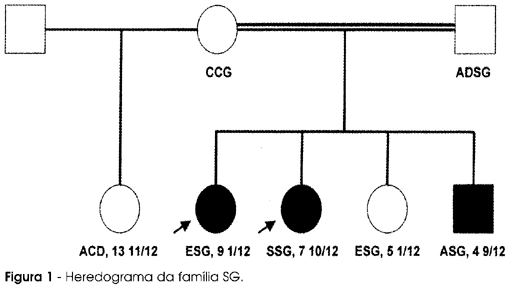
Descrevemos uma família (SG), acompanhada no ambulatório do Dr. Daniel Giannella-Neto, que apresenta três indivíduos portadores de deficiência combinada de hormônios hipofisários secundária a mutação no gene Prop-1. Conforme demonstrado na figura 1, os pacientes afetados incluem duas irmãs e 1 irmão filhos de pais consangüíneos (primos de primeiro grau).
Avaliação Clínica e Hormonal
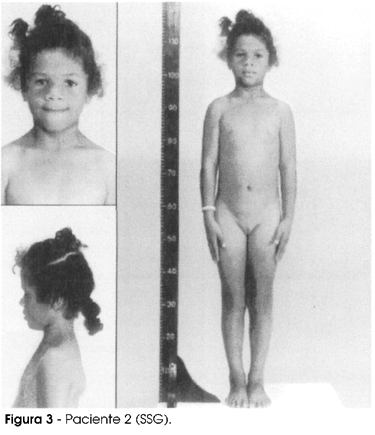
Os três pacientes nasceram de parto normal, a termo, hospitalar, em apresentação cefálica, sem intercorrências neo e pós-natais, apresentando DNPM normal. Ao exame físico, além do importante déficit pondo-estatural, apresentavam facies de boneca, voz de timbre agudo e aumento da gordura abdominal. A inteligência é normal. As pacientes 1 e 2 (fotografadas quando já estavam em vigência do tratamento) são mostradas nas figuras 2 e 3, respectivamente.
Os parâmetros de crescimento e os dados laboratoriais são apresentados na tabela 1.
Ensaios Hormonais
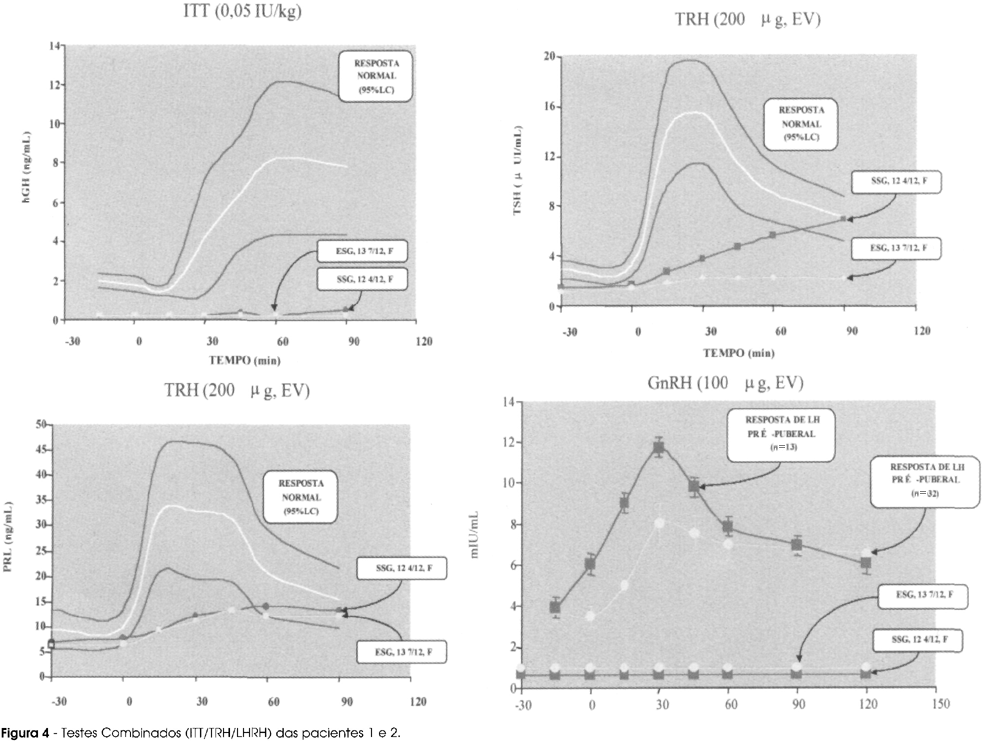
Os pacientes foram submetidos a um Teste Combinado para avaliação do eixo hipotálamo-hipofisário através da administração endovenosa de insulina (0,05 U/kg), TRH (200mg) e LHRH (100m,g). As concentrações séricas dos diversos hormônios foram determinadas pelos métodos que se seguem: GH, IGF-I e IGFBP-3 por ensaio imunoradiométrico (IRMA), LH e FSH por ensaio imunofluorimétrico, T3, T4 e TSH por ensaio imunoenzimétrico, PRL e estradiol por ensaio fluorimétrico e cortisol e DHEAS por radioimunoensaio (RIA).
Os Testes Combinados das pacientes 1 e 2 são mostrados na figura 4, enquanto o Teste descontinuado do paciente 3 é apresentado na tabela 2.
Avaliação Radiológica (RNM)
Paciente 1. (em 12/94): Formação expansiva intra-selar com expansão supra-selar, de contornos regulares, com intensidade de sinal finamente heterogênea em T1, sem realce pós-contraste, medindo aproximadamente 0,9cm de diâmetro. Haste hipofisária afilada e centrada. Imagem sugestiva de microadenoma hipofisário. (em 11/98): Hipófise de tamanho normal. A direita, observa-se área que não capta contraste, sugestiva de microadenoma hipofisário (figura 5).
Paciente 2. (em 12/94): Hipófise diminuída de tamanho, afilamento da haste hipofisária. 11/98: Normal (figura 6).
Paciente 3. (em 11/98): Aumento do tamanho hipofisário (glândula abaulada), intensidade de sinal homogênea, sem realce pós-contraste (figura 7).
Análise Molecular
Após extração do DNA genômico a partir de leucócitos de sangue periférico, procedeu-se à amplificação do exon 2 do gene Prop-1 por PCR e posterior seqüenciamento automático (figura 8) dos fragmentos amplificados. Os 3 indivíduos estudados apresentaram-se homozigotos para a deleção dinucleotídica A301G302, enquanto seus pais foram heterozigotos para a mesma mutação, caracterizando um padrão de herança autossômica recessiva.
Evolução
Os três pacientes afetados estão recebendo hormônio tireoidiano e GH (0,1 U/kg/dia). A curva de crescimento antes e após a instituição da terapêutica de substituição hormonal é apresentada na figura 9.
COMENTÁRIOS
O que chamou a atenção nestes casos, e nos motivou a iniciar a investigação do ponto de vista molecular, foi a incidência familiar da deficiência combinada de hormônios hipofisários (GH, PRL, TSH e gonadotrofinas) associada ao aumento do volume hipofisário com imagens sugestivas de microadenoma. O DNA desta família foi enviado ao Dr. John Parks, do Departamento de Pediatria da Emory University (Atlanta, Georgia) que estava justamente estudando famílias com características semelhantes em colaboração com o grupo do Dr. Rosenfeld, que havia descrito a mutação do gene Prop-1 nos camundongos Ames e estava estudando mutações deste gene em famílias portadoras de hipopituitarismo.
Ainda permanecem obscuras as razões para o aumento hipofisário observado em alguns indivíduos portadores de mutações no gene Prop-1. Uma paciente acompanhada pelo Dr. Parks foi submetida à cirurgia por apresentar importante expansão supra-selar. O exame anátomo-patológico revelou apenas a presença de material amorfo e ausência de células. É importante ressaltar que o volume hipofisário pode modificar-se durante a evolução destes pacientes, como foi observado na paciente 1 aqui apresentada.
REVISÃO DO TEMA
A adenohipófise deriva da bolsa de Rathke, uma invaginação da ectoderme estomodeal que surge no 8,5° de vida embrionária (E8,5) do Camundongo, enquanto a neurohipófise origina-se da ectoderme neural na base do diencéfalo. Existem evidências de que a adenohipófise também apresenta origem neuroectodérmica, derivando da porção mais anterior da placa neural (1,2). O desenvolvimento da adenohipófise está intimamente relacionado a sinais gerados pelo diencéfalo em desenvolvimento (3) (que irá originar o hipotálamo), assim como a uma cascata de fatores de transcrição tecido-específico que regulam a diferenciação e proliferação celular (4).
Precedendo a formação da bolsa de Rathke, genes homeobox são expressos, determinando o desenvolvimento de grupos celulares específicos em hipófise anterior (5). Estes genes codificam um domínio de 60 aminoácidos (homeo domain) presente em inúmeras proteínas de eucariotos envolvidas na regulação do desenvolvimento, e têm seu nome derivado da identificação original no loci homeótico da Drosophila (genes que determinam a igualdade das estruturas corpóreas). O domínio homeo tem a capacidade de se ligar ao DNA, e é encontrado em fatores de transcrição. Six3 (6) e Rpx (7) (Rathke's pouch homeobox; também conhecido como Hesxl) são inicialmente expressos na placa neural anterior (por volta de E6,5). Enquanto Six3 continua a ser expresso na bolsa de Rathke e em outras regiões, a expressão de Rpx torna-se .restrita à bolsa de Rathke até aproximadamente E14,5, sendo que a diminuição de sua expressão coincide com o aparecimento dos tireotrofos, somatotrofos e lactotrófos. Mutações no gene Rpx foram descritas em associação com displasia septo-óptica em camundongos e seres humanos. Dois irmãos portadores de panhipopituitarismo, hipoplasia de nervo óptico, agenesia de corpo caloso e septo pelúcido são homozigotos para uma mutação no domínio homeo de Rpx, causando diminuição de sua capacidade de ligação ao DNA. Estes achados sugerem que este fator de transcrição desempenhe um papel crítico no desenvolvimento da hipófise, encéfalo e linha média (8).
Um fator de transcrição que contém um domínio homeo foi simultaneamente identificado por dois laboratórios e nomeado pituitary homeobox 1 (Ptx1) (9) e pituitary OTX-related factor (P-OTX) (10). O Ptx1 é expresso no estomodeo (E8,5) e estruturas derivadas, incluindo a bolsa de Rathke. Durante a fase de diferenciação e proliferação celular hipofisária (E10,5-E15), é expresso nos lobos anterior e intermediário, agindo em associação com fatores de transcrição restritos a certas linhagens celulares para estimular a expressão de genes linhagem-específicos. Ptx1 age sinergisticamente com NeuroD1 para determinar a transcrição do POMC em corticotrofos (11), com Pit-1 para promover a expressão do gene da PRL (10) e com o receptor nuclear órfão SF-1 (steroidogenic factor-1) para ativar o promotor da cadeia b do LH. Além destas importante interações, Ptx1 também é necessário para a expressão do gene Lbx3, sugerindo um papel deste fator de transcrição nas etapas iniciais do desenvolvimento hipofisário (11). A expressão de Ptx1 persiste nas células corticotróficas da glândula adulta (9).
O gene homeobox Lbx3 é expresso simultaneamente à formação da bolsa de Rathke's (E9), persistindo durante o desenvolvimento da adenohipófise (12). Na ausência de expressão deste fator de transcrição, existe a formação da bolsa de Rathke, sem, no entanto, ocorrer uma proliferação celular normal, resultando na falta das linhagens celulares hipofisárias, com exceção de um pequeno número de corticotrofos (13). O Lbx3 ativa o promotor da cadeia a (LH, ESH e TSH), atua sinergisticamente com o Pit-1 na ativação da transcrição da PRL e cadeia b do TSH e é necessário para a expressão contínua do Rpx. Especula-se que este fator de transcrição participe da manutenção de algumas linhagens celulares hipofisárias, pois está presente na glândula madura (14).
A mutação homozigota de Lhx4, outro fator de transcrição que contém um domínio homeo, não afeta a formação da bolsa de Rathke, mas interfere com a proliferação de tipos celulares específicos, induzindo uma diminuição significativa no número de somatotrofos e lactotrófos (15). Os camundongos mutantes (homozigotos) para os genes Lhx3 e Lhx4 apresentam uma interrupção no desenvolvimento hipofisário mais precoce e mais severo que a alteração observada nos mutantes para apenas um dos genes. Embora a expressão sobreposta destes dois fatores de transcrição sugira uma redundância funcional, Lhx3 é mais importante que Lhx4 no processo de maturação hipofisária (16). O fator de transcrição Pit-1 começa a ser expresso ao redor de E13,5 (17), sendo crítico para a diferenciação e sobrevivência dos somatotrofos, lactotrófos e tireotrofos, assim como para a transcrição dos genes codificadores de GH, PRL e cadeia b do TSH. Mutações 110 gene do Pit-l são responsáveis pela deficiência combinada de hormônios hipofisários observada nos camundongos anões Snell e Jackson, que apresentam hipoplasia pituitária anterior secundária à ausência das três linhagens celulares dependentes do gene Pit-1 (18). Diferentes mutações neste gene têm sido descritas em seres humanos portadores de nanismo associado a deficiência de GH, PRL e TSH (19).
Gene Prop-1 (Prophet of Pit-1)
Os camundongos anões Ames apresentam um fenótipo quase idêntico ao dos camundongos Snell e Jackson, com severa redução no número de somatotrofos, lactotrófos e tireotrofos devido a deficiência de ativação do gene do Pit-1 durante a ontogênese hipofisária (20). Sornson et al, identificaram no cromossoma 11 o gene mutado nos camundongos Ames, e o nomearam Profeta de Pit-1 (Prop-1), uma vez que codifica um fator de transcrição hipófise-específico necessário para a expressão do gene Pit-1. A seqüência aberta de leitura do gene Prop-1 murino prediz uma proteína de 223 aminoácidos que contém um domínio homeo simile composto de três helices capazes de se ligarem ao DNA (4). A organização genômica do Prop-1 é similar entre o Camundongo e o homem (21), com a região codificadora apresentando 78% de homologia. A região mais conservada compreende a seqüência codificadora do domínio homeo (22), sendo as demais regiões menos conservadas (21). O gene humano está localizado no cromossoma 5 (23) (região q35), sendo composto de pelo menos 3 exons e 2 introns que ocupam um segmento genômico de aproximadamente 4kb. As áreas de transição intronexon contêm seqüências consenso (GT-AG) (22) que participam do mecanismo de splicing durante o processamento do transcrito primário de RNA mensageiro (retirada das regiões correspondentes aos introns, de forma que apenas as seqüências correspondentes aos exons estejam presentes no RNA mensageiro maduro).
O estudo dos efeitos do Prop-1 sobre a expressão do gene Pit-1 sugere uma de duas possibilidades: ou o Prop-1 exerce efeitos indiretos sobre a transcrição do Pit-1, ou exerce efeitos diretos em associação com outros fatores de transcrição, não sendo capaz de atuar isoladamente (4),
A expressão do gene Prop-1 é observada na região dorsal da adenohipófise em desenvolvimento por volta do E10-E10.5, alcançando expressão máxima um dia e meio (E12) antes do início da expressão do Pit-1 na área caudomedial, onde as linhagens celulares dependentes de Pit-1 irão desenvolver-se. A expressão de Prop-1 é detectada até E14,5, quando declina significativamente, não estando presente na glândula madura. No camundongo Ames, o desenvolvimento hipofisário não é afetado até E13-E13,5, quando anormalidades no padrão de distribuição celular tornam-se evidentes e células que normalmente expressam Pit-1 não se acumulam na área caudomedial. As células que se originam antes de E13 - corticotrofos, gonadotrofos e tireotrofos da região rostral- estão normais (4). A adenohipófise madura é hipocelular, contendo apenas raros aglomerados dos tipos celulares dependentes de Pit-1 (24) e algumas células que expressam a-MSH atipicamente localizadas neste lobo (4).
Embora a dismorfogênese hipofisária observada no camundongo Ames esteja associada a perda de ativação do gene Pit-1, existem algumas diferenças entre os camundongos Ames e os Snell. Em contraste com os primeiros (20,24), os últimos não apresentam nenhuma célula que expresse GH, PRL ou TSH. A presença de algumas destas células na pituitária do camundongo Ames pode indicar atividade remanescente da proteína Prop-1 mutada ou autonomia parcial da expressão do gene Pit-1 (25). Adicionalmente, as alterações no desenvolvimento da bolsa de Rathke observadas nos camundongo Ames são detectadas antes mesmo do início da expressão do Pit-1 (26).
Os camundongos Ames apresentam tamanho normal ao nascimento, seguindo-se um severo retardo de crescimento (27). Além do déficit de GH, PRL e TSH, exibem níveis plasmáticos diminuídos de gonadotrofinas. Tang et al. detectaram uma redução no conteúdo de gonadotrofinas e de RNA mensageiro de cadeia b, assim como na liberação de gonadotrofinas pela adenohipófise (28). Uma vez que os camundongos Snell apresentam expressão normal de gonadotrofinas, parece improvável que o defeito no setor gonadotrófico observado nos camundongos Ames seja secundário à ausência de células dependentes de Pit-1 terminalmente diferenciadas, sendo possível que o gene Prop-1 esteja envolvido na determinação dos gonadotrofos através dos precursores das células dependentes de Pit-1 originados antes da diferenciação terminal (4).
A mutação responsável pelo fenótipo Ames foi detectada na região que codifica a primeira a-hélice do domínio homeo, onde uma transição T C determina a substituição de uma serina por uma prolina na posição 83 (S83P). Esta troca de aminoácido diminui a afinidade da proteína Prop-1 pelo DNA, interferindo em sua habilidade de se ligar ao DNA e ativar a transcrição. Três outras modificações na seqüência de aminoácidos foram descritas no domínio carboxi-terminal do Prop-1 de camundongos Ames (G155A, S171A e a inserção de uma prolina entre as posições 208 e 209), caracterizando polimorfismos e não mutações, uma vez que não causam qualquer repercussão funcional (4).
C determina a substituição de uma serina por uma prolina na posição 83 (S83P). Esta troca de aminoácido diminui a afinidade da proteína Prop-1 pelo DNA, interferindo em sua habilidade de se ligar ao DNA e ativar a transcrição. Três outras modificações na seqüência de aminoácidos foram descritas no domínio carboxi-terminal do Prop-1 de camundongos Ames (G155A, S171A e a inserção de uma prolina entre as posições 208 e 209), caracterizando polimorfismos e não mutações, uma vez que não causam qualquer repercussão funcional (4).
Wu et al. (21) foram os primeiros a descrever pacientes portadores de subtipos de deficiência combinada de hormônios hipofisários (envolvendo GH, TSH, PRL e gonadotrofinas) secundariamente a mutações inativadoras do gene Prop-1. Irmãos afetados em duas famílias foram homozigotos para uma deleção dinucleotídica na seqüência GAGAGAG no exon 2, originando uma mudança no quadro de leitura a partir do codon 101 e determinando um stop codon prematuro na posição 109. A proteína resultante não apresenta o domínio homeo de ligação ao DNA e o domínio carboxi terminal de transativação. Os membros afetados da terceira família descrita apresentaram-se homozigotos para uma transição CT no codon 120, levando a uma mutação missense (ArgininaCisteína) situada na terceira hélice do domínio homeo (R120C). O indivíduo afetado da quarta família foi caracterizado como um heterozigoto composto para a deleção dinucleotídica citada anteriormente e para uma transversão TA no codon 117, causando uma mutação missense (FenilalaninawInterleucina) localizada na terceira hélice do domínio homeo (F117I). Os estudos funcionais revelaram que os produtos protéicos das mutações R120C e F117I apresentam redução da afinidade ao DNA e da capacidade de ativar a transcrição, enquanto a proteína truncada derivada da deleção dinucleotídica não se liga ao DNA e não é capaz de induzir transcrição, caracterizando uma mutação com total ausência de função (functional null mutation) do gene Prop-1.
A deleção dinucleotídica parece ser a mutação do gene Prop-1 mais freqüentemente encontrada, tendo sido descrita em pacientes originados dos EUA, Jamaica (29), Rússia (30,31), Turquia (22,32), Brasil, Europa (32) e República Dominicana (33). A seqüência GAGAGAG (do nucleotídeo 296 a 302) tem sido considerada um hot spot para mutações, susceptível a desalinhamentos do DNA durante o crossing over e deleções (32).Uma vez que não é possível determinar-se qual nucleotídeo foi excluído desta seqüência (pois qualquer combinação de deleções GA ou AG gera os mesmos resultados), esta mutação tem sido chamada de A301G302 (21) ou de 296 delGA (30). Um outro hot spot para mutações tem sido reportado no exon 2, consistindo de uma deleção dinucleotídica (149delGA) que cria uma mudança no quadro de leitura a partir do codon 51 e determina o aparecimento de um stop codon prematuro na posição 109. Esta mutação foi descrita em famílias russas com deficiência combinada de hormônios hipofisários, estando presente em heterozigose composta com a mutação A301G302 (31).
Duquesnoy et al. (22) descreveram uma transição CT no codon 73 (exon 2), responsável por uma mutação missense (R73C) na região amino-terminal do domínio homeo em uma família da Tunísia. Este grupo também relatou uma mutação intrônica (AT) no sítio de splicing que antecede o exon 3 em uma família egípcia. Esta mutação impede um processo de splicing normal, originando um transcrito de RNA mensageiro que conserva o intron 2 e outro resultante da utilização de um sítio críptico de splicing contido no exon 3, o qual perde 12 nucleotídeos.
DISCUSSÃO ABERTA
Dr. Geraldo Medeiros-Neto (Professor Associado da Disciplina de Endocrinologia e Metabologia da Faculdade de Medicina da USP):
Nós temos uma família que está sendo estudada em Minas Gerais com 4 indivíduos afetados em uma irmandade de 11. O mais marcante nesta família é que o ACTH está conservado, como se espera, enquanto e evolução da função tireoidiana foi muito curiosa pois, conforme você também observou em uma das pacientes, existe resposta ao TRH, e eles somente desenvolveram hipotireoidismo tardiamente. Aparentemente, o tireotrofocito está resistindo. Existe alguma explicação para isto?
Dra. Maria Lúcia Giannella:
Com a descrição de um maior número de casos, é muito provável que possamos observar certa heterogeneidade nas manifestações clínicas de pacientes portadores de mutações no gene Prop-1, ou ainda modificações na apresentação clínica que aconteçam durante a evolução. Nesta família aqui descrita, o irmão mais novo apresentou liberação de PRL, ao contrário de suas irmãs diagnosticadas em idade mais avançada. É provável também que o quadro clínico varie conforme o tipo de mutação. Assim, mutações mais severas, como a A301G302 e a 149 delGA, que em estudos in vitro abolem completamente a função da proteína Prop-1, devem determinar quadros mais severos que outras mutações em que ainda observa-se função remanescente deste fator de transcrição, como as mutações missense R120C ou E117I.
Dr. Geraldo Medeiros-Neto:
Na semana passada, recebemos a confirmação de que esta família de Minas Gerais apresenta justamente a mutação A301G302.
Dra. Maria Lúcia Giannella:
Acredito que o estudo de um maior número de pacientes portadores de mutações no gene Prop-1 poderá nos trazer maiores informações sobre as variáveis (tipo de mutação, evolução do quadro com a idade) que influenciam a apresentação clínica.
Dr. Marcello Delano Bronstein (Professor Livre Docente de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP):
Começando pela parte clínica: o paciente 3, apesar de ter uma PRL basal relativamente alta, tem uma resposta diminuída.
Dra. Maria Lúcia Giannella:
O Teste Combinado realizado neste paciente somente consta dos tempos -15, 0 e 15 minutos, pois houve perda da veia, portanto, não sei se podemos dizer que já houve resposta.
Dr. Marcello Delano Bronstein:
Caso esta resposta estivesse em um tempo mais tardio, seria diminuída. Mesmo em 15 minutos, já se inicia a elevação da PRL, sendo o pico entre 15 e 30 minutos. Como o pico mínimo é considerado 3 vezes o basal, poderia considerar-se a resposta diminuída; mas, de qualquer forma, você somente tem o tempo 15. As RNM são interessantes e na paciente 2 eu levantaria a suspeita de um cisto devido ao hipossinal com pequena captação periférica, já que isto pode ser descrito em hipopituitarismos congênitos.
Dra. Maria Lúcia Giannella:
Esta não foi nossa suspeita, pois esta imagem foi vista na segunda RNM, sendo que na primeira a imagem era bastante diferente, inclusive sugestiva de microadenoma e muito semelhante à imagem vista na RNM apresentada para o paciente 3. Portanto, houve uma alteração da imagem hipofisária na evolução deste caso, o que já foi observado para outros pacientes com aumento do volume hipofisário concomitante à mutação no gene Prop-1.
Dr. Marcello Delano Bronstein:
No caso do paciente 3, eu diria que seguramente não está diminuída, mas é uma hipófise no limite. Existe um pequeno abaulamento superior, mas é difícil classificar como aumento.
Falando agora de aspectos mais básicos, é curioso observar que na deficiência de Pit-1 existe acometimento de GH e PRL, que são derivados da mesma célula-mãe, e aqui você também tem acometimento do TSH, que é uma glicoproteína.
Dra. Maria Lúcia Giannella:
Tanto mutações no gene Pit-1 como no Prop-1 cursam com acometimento dos setores hipofisários produtores de GH, PRL e TSH, e, no caso do Prop-1, também de gonadotrofinas. O fato de a deficiência de um único fator de transcrição (Pit-1) acometer 3 linhagens celulares é sugestivo de que estas 3 linhagens originem-se de uma mesma célula precursora.
Dr. Marcello Delano Bronstein:
E porquê não haveriam manifestações nos setores gonadotrófico e adrenocorticotrófico?
Dra. Maria Lúcia Giannella:
Existe uma cascata de fatores de transcrição e a ativação seqüencial de cada um deles é responsável pelo desenvolvimento e proliferação das diversas linhagens celulares. Quanto mais inicial na cascata, maior a possibilidade daquele fator de transcrição interferir com um maior número de linhagens. Assim, por exemplo, a inativação de Lhx3 interfere com a formação de gonado, tireo, lacto e somatotrofos. Já os corticotrofos são mais dependentes de um outro fator, o Ptx-1 (ou POTX). É importante ressaltar, porém, que cada fator de transcrição não age isoladamente, mas em conjunto com outros fatores de transcrição.
Dr. Marcello Delano Bronstein:
No caso de mutações no gene Prop-1, existe a possibilidade de as manifestações dependerem apenas da não ativação do gene Pit-1?
Dra. Maria Lúcia Giannella:
O mecanismo de ação do Prop-1 não está completamente esclarecido. Sabe-se, por estudos in vitro, que ele não atua isoladamente na ativação do gene Pit-1. A inativação do gene Pit-1 não cursa com deficiência de gonadotrofinas, ao contrário do que ocorre com a inativação do gene Prop-1, portanto, é muito provável que o quadro clínico observado nas mutações do gene Prop-1 não dependa exclusivamente da falta de ativação do gene Pit-1.
Dr. Marcello Delano Bronstein:
Se eu não me engano, o Prof. Dr. Sérgio Toledo descreveu alguns pacientes que também teriam suspeita de deficiência corticotrófica.
Dra. Maria Lúcia: Não sei se estes pacientes têm mutação no gene Prop-1.
Dr. Sérgio Pereira de Almeida Toledo (Professor Associado da Disciplina de Endocrinologia e Metabologia da Faculdade de Medicina da USP):
Aquela família tem 14 ou 15 membros e nós ainda não estudamos todos, porém, observamos que alguns pacientes têm deficiência e outros não. Inicialmente imaginávamos que isto era ligado ao fator idade mas, internando quatro pacientes recentemente (dois homens e duas mulheres) com mais de 40 anos, observou-se que dois apresentavam deficiência e dois não.
Dra. Maria Lúcia Giannella:
Já se sabe qual o gene mutado nestes pacientes?
Dr. Sérgio Pereira de Almeida Toledo:
Encontra-se, ainda, sob investigação.
Dr. Ivo Jorge Prado Arnhold (Professor Livre Docente de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP):
Quanto à nomenclatura, se em relação à deleção de nucleotídeos raciocinarmos no sentido da leitura do gene (5' para 3'), qualquer que seja a dupla de nucleotídeos deletada, o que vai faltar são os últimos dois. Assim, optamos por chamar a mutação de 301/302, como foi chamada por Wu na primeira descrição. Acho que isto é importante para uniformizar a nomenclatura.
Dra. Maria Lúcia Giannella:
Eu também tenho me referido a esta mutação como A301/G302, mas acho importante que as pessoas fiquem atentas, pois vão encontrar a mesma mutação com diferentes nomes (296, 97; AG296).
Dr. Ivo Jorge Prado Arnhold:
Na RNM que você mostrou no Congresso Brasileiro, chamou a atenção o hipersinal em uma das pacientes. Em uma paciente acompanhada por nosso grupo, a RNM aos 8 anos evidenciava um hipersinal em T1 e sela aumentada. Oito anos depois, o parênquima hipofisário havia colabado e o hipersinal havia desaparecido. Assim, este hipersinal poderia chamar a atenção do radiologista para um diagnóstico de mutação no gene Prop-1.
Sobre o aparecimento mais tardio das deficiências, está claro que pode ser evolutivo nas diferentes mutações que foram apresentadas. No trabalho do grupo do Dr. Mullis que foi publicado no JCEM de outubro de 1998, eles demonstraram deficiência de GH com resposta até 8 a 10ng/mL, e que com o tempo foi diminuindo. Uma das nossas pacientes e uma relatada pelo Dr. Parks apresentam deficiência de cortisol, ou pelo menos uma diminuição da reserva corticotrófica com o passar do tempo, de forma que esta deficiência poderia fazer parte do quadro clínico.
Dra. Maria Lúcia Giannella:
Nos casos que eu levantei na literatura, não havia relatos de deficiência de cortisol.
Dr. Ivo Jorge Prado Arnhold:
O Dr. Parks fala de pacientes com deficiência de cortisol no abstract apresentado no Endocrine Society.
Dra. Berenice Bilharinho de Mendonça (Professora Titular de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP):
Eu queria fazer um comentário a respeito do que o Dr. Ivo falou. Realmente existem fatores que trabalham em conjunto com o Prop-1, pois, conforme mencionado no trabalho do Dr. Mullis, a própria deficiência de GH pode ser tardia, e se nós sabemos que a expressão de Prop-1 é transitória no período embrionário, o que é que mantém este somatotrofo? Depois, ele deixa de funcionar; assim, seguramente existem genes atuantes que não agem sozinhos, e que temos que buscar. Eu queria deixar uma mensagem para todos que trabalham com déficit de GH: este aspecto de sela grande em pacientes com deficiência de GH, é uma coisa que há anos nos chama a atenção. Nós víamos estes pacientes e não sabíamos o que eles tinham, mas era esquisito, pois normalmente panhipopituitarismo tem sela pequena. Quando não tínhamos a RNM, falávamos que se tratava de sela vazia. Após a RNM, vimos que este não era o caso. Nós temos condições de fazer o screening para esta mutação (A301/G302), que é a mais freqüente. Aqui entra outra discussão, porque no comprometimento do gene Prop-1 você tem resposta de deficiências parciais dependendo da mutação, e a diferença entre o que é hipofisário e o que é hipotalâmico fica muito complicada. Este é o trabalho da Geralda, que é orientanda do Ivo, a relação entre a RNM, as secreções hormonais e o estudo do gene. Isto é muito importante, e vai nos chamar a atenção para vários outros genes, inclusive de diferenciação hipotalâmica.
Dra. Maria Lúcia Giannela:
Eu imagino que a partir de agora, cada vez mais mutações nestes fatores de transcrição envolvidos na ontogênese hipotálamo-hipofisária passarão a ser descritas. Eu não tenho muitas informações a respeito dos fatores de transcrição atuantes no desenvolvimento hipotalâmico. O Six3, Pax e Rpx são expressos em algumas regiões cerebrais, inclusive no hipotálamo. Alguns deles permanecem expressos, outros, como o Rpx, ficam restritos à bolsa de Rathke. Sabe-se que existem inter-relações entre os fatores de transcrição. Assim, o próprio camundongo Ames apresenta um aumento na expressão de um fator relacionado ao desenvolvimento do tubo neural, o Brn4. A razão para este aumento não é conhecida.
Dra. Maria Adelaide Albergaria Pereira (Médica Assistente Doutora do Serviço de Endocrinologia do Hospital das Clínicas da Faculdade de Medicina da USP):
O aumento do volume hipofisário também pode ser encontrado na mutação do gene Pit-1?
Dra. Maria Lúcia Giannella:
Não, somente no acometimento do gene Prop-1. Acredita-se que o fenótipo encontrado na mutação do gene Prop-1 decorra basicamente da falta de ativação do gene Pit-1, no entanto, nas mutações deste último gene, não se observa esta alteração na morfologia hipofisária. Embora isto não seja mencionado na literatura, eu acredito que o Prop-1 possa ter algum outro papel na ontogênese hipofisária, independente do Pit-1, que justifique o aumento do volume hipofisário encontrado em alguns casos.
REFERÊNCIAS
1. Eagleson GW, Harris WA. Mapping of the presumptive brain regions in the neural plate of Xenopus laevis. J Neurobiol 1990;21:427-40.
2. Couly G, Le Douarin NM. The fate map of the cephalic neural primordium at the presomitic to the 3-somite stage in the avian embryo. Development 1988; 103(suppl):101-13.
3. Watanabe YG. Effects of brain and mesenchyma upon the cytogenesis of rat adenohypophysis in vitro. Cell Tissue Res 1982;227:257-66.
4. Sornson, et al. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature 1996;384:327-33.
5. Treiner M, Rosenfeld MG. The hypothalamic-pituitary axis: co-development of two organs. Curr Opin Cell Biol 1996;8:833-43.
6. Olivier G, Wehr R, Jenkins NA, et al. Homeobox genes and connective tissue patterning. Development 1995;121:693-705.
7. Hermesz E, Mackem S, Mahon K. Rpx: a novel anterior-restricted homeobox gene progressively activated in the prechordal plate, anterior neural plate and Rathke's pouch of the mouse embryo. Development 1996;122:41-52.
8. Dattani MT, et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nature Genetics 1998;19:125-33.
9. Lamonerie T, Tremblay JJ, Lanctôt C, Therrien M, Gauthier Y, Drouin J, Ptx-1, a bicoid-related homeobox transcription factor involved in transcription of the pro-opi-omelanocortin gene. Genes Dev 1996;10:1284-95.
10. Szeto DP, Ryan AK, O'Connell SM, Rosenfeld MG. P-OTX: a Pit-1 interacting homeodomain factor expressed during anterior pituitary gland development. Proc Natl Acad Sci USA 1996;93:7706-10.
11. Drouin J, Lamolet B, LamonerieT, Lanctôt C, Tremblay JJ. The Ptx family of homeodomain transcription factors during pituitary developments. Mol Cell Endocrinol 1998; 140:31-6.
12. Zhadanov AB, Bertuzzi S, Taira M, Dawid IB, Westphal H. Expression pattern of the murine UM class homeobox gene LHX3 in subsets of neural and neuroendocrine tissues. Dev Dyn 1995;202:354-64.
13. Sheng HZ, Zhadanov AB, Mosinger B Jr, et al. Specifications of pituitary cell lineages by the UM homeobox gene Lhx3. Science 1996;272:1004-7.
14. Bach I, Rhodes SJ, Pearse RV II, et al. P-Lim, a UM homeodomain factor, is expressed during pituitary organ and cell commitment and synergizes with Pit-1. Proc Natl Acad Sci USA 1995;92:2720-4.
15. Flavell DM, Wells T, Wells SE, Carmignac DF, Thomas GB, Robinson IC. Dominant dwarfism in transgenic rats by targeting human growth hormone (GH) expression to hypothalamic GH-releasing factor neurons. EMBO J 1996;15:3871-9.
16. Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, Westphal H. Multistep control of pituitary organogenesis. Science 1997;278:1809-12.
17. Andersen B, Rosenfeld MG. Pit-1 determines cell types during development of the anterior pituitary gland. J Biol Chem 1994;269:29335-8.
18. Li S, Crenshaw EBI, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. Dwarf locus mutants, which lack three pituitary cell types, result from mutations in the POU domain gene, Pit-1. Nature 1990;347:528-33.
19. Cogan JD, Phillips JA III. Growth disorders caused by genetics defects in the growth hormone pathway. Adv Pediat 1998;45:338-61.
20. Andersen B, et al. The Ames dwarf gene is required for Pit-1 gene activation. Develop Biol 1995;172:495-503.
21. Wu W, et al. Mutations in Prop-1 cause familial combined pituitary hormone deficiency. Nature Genet 1998;18:147-9.
22. Duquesnoy P, et al. Human Prop-1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency. FEBS Letters 1998;437:216-20.
23. Rosenfeld MG, Wu W. Personal Communication. San Diego, CA, 1/23/1998.
24. Gage PJ, Roller ML, Saunders TL, Scarlett LM, Camper SA. Anterior pituitary cells defective in the cell-autonomous factor, df, undergo cell lineage specification but not expansion. Development 1996;122:151-60.
25. Parks JS, Adess ME, Brown MR, Genes regulating hypothalamic and pituitary development. Acta Paediatr Suppl 1997;423:28-32.
26. Gage PJ, Brinkmeier ML, Scarlett LM, Knapp LT, Camper SA, Mahon KA. The Ames dwarf gene, df, is required early in pituitary ontogeny for the extinction of Rpx transcription and initiation of lineage-specific cell proliferation. Mol Endocrinol 1996;10:1570-81.
27. Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the aging process. (Letter) Nature 1996;384:33.
28. Tang K, Bartke A, Gardiner CS, Wagner TE, Yun JS. Gonadotrophin secretion, synthesis and gene expression in human growth hormone transgenic mice and in Ames dwarf mice. Endocrinology 1993;132:2518-24.
29. Parks JS, et al. Natural history and molecular mechanisms of hypopituitarisms with large sella turcica. Proc Endocrine Soc 80th Ann Mtg New Orleans, LA, 1998, p471.
30. Fofanova OV, Takamura N, Kinoshita E, Parks JS, Brown MR, Peterkova VA, et al. A mutational hot spot in the Prop-1 gene in Russian children with combined pituitary hormone deficiency. Pituitary 1998;1:45-9.
31. Fofanova OV, Takamura N, Kinoshita E, Parks JS, Brown MR, Peterkova VA, et al. Compound heterozygous deletion of the Prop-1 gene in children with combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998;83:2601-4.
32. Cogan JD, et al. The Prop-1 2-base pair deletion is a common cause of combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998;83:3346-9.
33. Rosenblom AL, Almonte AS, Brown MR, Fisher DA, Baumbach L, Parks JS. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the Prop-1 gene. J Clin Endocrinol Metab 1999 (in press).
Endereço para correspondência:
Daniel Giannella Neto
Laboratório de Nutrição Humana e Doenças Metabólicas -
LIM 25B
Hospital das Clínicas da FMUSP
Av. Dr. Arnaldo, 455 - sala 4703
01246-903 São Paulo, SP
Esta seção é parte de um projeto de educação continuada em Endocrinologia e Metabologia e está sendo publicada graças ao patrocínio e à colaboração da

Responsáveis pela Apresentação e Discussão: Maria Lúcia C. C. Giannella e Daniel Giannella Neto Responsável pela Edição: Maria Adelaide Albergaria Pereira
Local e Data da Reunião: Serviço de Endocrinologia e Metabologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo - 10/12/98
Datas de Publicação
-
Publicação nesta coleção
04 Out 2005 -
Data do Fascículo
Abr 2000