Abstracts
OBJECTIVE: To search for mutations in DAX1/NR0B1A gene in siblings to establish the molecular etiology of the adrenal hypoplasia congenita (AHC), a rare potentially life-threatening disorder. CASE REPORT: We describe two siblings who presented with salt-wasting syndrome in the newborn period and received hormonal replacement for primary adrenal insufficiency. A diagnostic hypothesis of AHC was suspected because the children maintained, during hormonal treatment, low plasma 17-OH progesterone (17-OHP) and androgens, despite high ACTH levels. RESULTS: DAX1 gene was studied by molecular analysis, which showed a mutation, confirming the diagnosis in the siblings and a heterozygous state in the mother. Direct sequencing of DAX1 revealed an insertion of an adenine base (c1382-1383 A ins), which lead to a pMet461Asp substitution. CONCLUSION: A novel frameshift mutation of DAX1 gene, which established the molecular etiology of the AHC in the siblings, was identified. Obtaining a precise genetic diagnosis of this adrenal disorder, which, sometimes, cannot be confirmed only by clinical aspects, may have important implications for the long-term management of the disease.
Congenital; Congenital; Congenital; adrenal insufficiency; frameshift mutation; genetic counseling
OBJETIVO: Pesquisar mutações no gene DAX1/NR0B1A em dois irmãos com suspeita de hipoplasia adrenal congênita (HAC), rara doença potencialmente fatal, para estabelecer sua etiologia molecular. RELATO DOS CASOS: São apresentados os relatos de dois irmãos com síndrome perdedora de sal no período neonatal que receberam terapia de reposição hormonal para insuficiência adrenal primária. O diagnóstico de HAC foi suspeitado porque as crianças mantiveram, durante o tratamento hormonal, níveis plasmáticos reduzidos de 17-OH-progesterona e andrógenos ao lado de níveis elevados de ACTH. RESULTADOS: A análise molecular do gene DAX1 mostrou a mutação, confirmando o diagnóstico nos irmãos e o estado heterozigoto da mãe. No sequenciamento direto do DAX1 foi encontrada inserção de uma adenina (c1382-1383 A ins), levando à substituição pMet461Asp. CONCLUSÃO: Uma nova mutação da fase de leitura no gene DAX1 foi identificada, estabelecendo a etiologia molecular da HAC nos dois irmãos. Um diagnóstico genético preciso deste distúrbio adrenal, frequentemente não confirmado apenas pelos aspectos clínicos, pode ter importantes implicações para o manuseio em longo prazo da doença.
Doenças congênitas; Doenças congênitas; Doenças congênitas; insuficiência adrenal; mutação da fase de leitura; aconselhamento genético
CLINICAL CASE REPORT
Identification of a novel mutation in DAX1/NR0B1A gene in two siblings with severe clinical presentation of adrenal hypoplasia congenita
Identificação de nova mutação no gene DAX1/NR0B1A em dois irmãos com forma clínica grave de hipoplasia adrenal congênita
Rafael Machado Mantovani; Isabela Leite Pezzuti; Vera Maria Alves Dias; Ivani Novato Silva
Divisão de Endocrinologia Pediátrica, Departamento de Pediatria, Hospital das Clínicas, Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, MG, Brasil
Correspondence to Correspondence to: Ivani Novato Silva Departamento de Pediatria Faculdade de Medicina, UFMG Av. Alfredo Balena, 190, sala 267 ivanins@medicina.ufmg.br
ABSTRACT
OBJECTIVE: To search for mutations in DAX1/NR0B1A gene in siblings to establish the molecular etiology of the adrenal hypoplasia congenita (AHC), a rare potentially life-threatening disorder.
CASE REPORT: We describe two siblings who presented with salt-wasting syndrome in the newborn period and received hormonal replacement for primary adrenal insufficiency. A diagnostic hypothesis of AHC was suspected because the children maintained, during hormonal treatment, low plasma 17-OH progesterone (17-OHP) and androgens, despite high ACTH levels.
RESULTS:DAX1 gene was studied by molecular analysis, which showed a mutation, confirming the diagnosis in the siblings and a heterozygous state in the mother. Direct sequencing of DAX1 revealed an insertion of an adenine base (c1382-1383 A ins), which lead to a pMet461Asp substitution.
CONCLUSION: A novel frameshift mutation of DAX1 gene, which established the molecular etiology of the AHC in the siblings, was identified. Obtaining a precise genetic diagnosis of this adrenal disorder, which, sometimes, cannot be confirmed only by clinical aspects, may have important implications for the long-term management of the disease.
Keywords: Congenital, hereditary, and neonatal diseases and abnormalities; adrenal insufficiency; frameshift mutation; genetic counseling
RESUMO
OBJETIVO: Pesquisar mutações no gene DAX1/NR0B1A em dois irmãos com suspeita de hipoplasia adrenal congênita (HAC), rara doença potencialmente fatal, para estabelecer sua etiologia molecular.
RELATO DOS CASOS: São apresentados os relatos de dois irmãos com síndrome perdedora de sal no período neonatal que receberam terapia de reposição hormonal para insuficiência adrenal primária. O diagnóstico de HAC foi suspeitado porque as crianças mantiveram, durante o tratamento hormonal, níveis plasmáticos reduzidos de 17-OH-progesterona e andrógenos ao lado de níveis elevados de ACTH.
RESULTADOS: A análise molecular do gene DAX1 mostrou a mutação, confirmando o diagnóstico nos irmãos e o estado heterozigoto da mãe. No sequenciamento direto do DAX1 foi encontrada inserção de uma adenina (c1382-1383 A ins), levando à substituição pMet461Asp.
CONCLUSÃO: Uma nova mutação da fase de leitura no gene DAX1 foi identificada, estabelecendo a etiologia molecular da HAC nos dois irmãos. Um diagnóstico genético preciso deste distúrbio adrenal, frequentemente não confirmado apenas pelos aspectos clínicos, pode ter importantes implicações para o manuseio em longo prazo da doença.
Descritores: Doenças congênitas, hereditárias e neonatais e anormalidades; insuficiência adrenal; mutação da fase de leitura; aconselhamento genético
BACKGROUND
Adrenal hypoplasia congenita (AHC) is a rare potentially life-threatening disorder of adrenal gland development. The condition can be inherited as an autosomal-recessive or X-linked disease, both forms presenting different adrenal morphologies (1). Its precise incidence is unknown, although it has been quoted as approximately 1:12,000 live births (2). However, some authors believe that the true incidence of the X-linked form of AHC is much lower and might occur anywhere between 1:140,000 and 1:1,200,000 children (3).
The X-linked form of AHC (OMIM: 300200) is caused by mutations of DAX1 gene (dosage-sensitive sex reversal, AHC, critical region on the X chromosome, gene 1), also called NR0B1 gene (nuclear receptor, NR, superfamily 0, group B, member 1). It results in primary adrenocortical failure due to the lack of the permanent adult adrenal cortical zone, replaced by large cytomegalic vacuolated eosinophilic cells (1).
DAX1 gene contains two exons of 1,168 and 245 bp, separated by a 3,385 bp intron (4,5). It is expressed in many tissues, like skin, breast and prostate, as well as at all levels of the hypothalamic-pituitary-adrenal and gonadal axis (6). DAX1 encodes a 470-amino acid protein, which contains a C-terminal region showing homology to the ligand-binding domains of other members of the NR superfamily, but that lacks the typical zinc finger DNA-binding domain at the N-terminal region. Instead, it contains cysteines, positioned in the same region, to form a potential novel zinc finger (1,7-9).
Until 2003, about 112 mutations in DAX1 gene had been described in patients with the X-linked form of AHC, most of them being frameshift mutations and small deletions (10). In the last few years, many other DAX1 mutations have been described elsewhere (11-18). There is a wide phenotypic expression of DAX1 deficiency, ranging from severe salt-wasting with glucocorticoid and mineralocorticoid insufficiency presented in early infancy to more insidious and progressive onset of symptoms later in childhood or even in adulthood (14,18-20).
Hypogonadotropic hypogonadism (HH) is also commonly associated with X-linked AHC, usually detected because of lack of pubertal development (21). It is caused by abnormalities in both hypothalamic and pituitary control of gonadotropin secretion (22). It has also been postulated that affected men may present an intrinsic defect in spermatogenesis, resulting in poor fertility prognosis compared with men with isolated idiopathic HH (23).
This variability of phenotypic expression of DAX1 deficiency underlines the importance of genetic counseling for known carrier mothers. Prenatal or early postnatal genetic analysis can identify asymptomatic affected males before occurrence of adrenal crisis, decreasing mortality risk and allowing adequate hormonal replacement (24,25).
The objective of this study was to search for mutations in DAX1 in two siblings to establish the molecular etiology of the AHC.
SUBJECTS AND METHODS
Case report
The proband was a term newborn, adequate for gestational age (weight: 2,820 g; length: 47 cm) without history of perinatal complications. At 30 days of life, he was admitted with vomiting, dehydration, failure to thrive and generalized skin hyperpigmentation. He presented with severe hyponatremia (Na+: 103 mEq/L) and normal potassium levels (K+: 3.6 mEq/L), which later on had increased (5.6 mEq/L). Plasma 17-OH progesterone (17-OHP) (440 ng/dL; RV: 53-186 ng/dL) and testosterone concentrations (520 ng/dL; RV: 6-496 ng/dL) were mildly elevated. Androstenedione levels (6.5 ng/mL; RV: <1.6 ng/mL) were found to be significantly elevated. Plasma ACTH levels were very high (2,437 pg/mL; RV: 12-70 pg/mL) as well as plasma renin activity (PRA) (27 ng/mL/h; RV: 0.3-1.6 ng/mL/h). The diagnosis of congenital adrenal hyperplasia, due to 21-OH deficiency (21-OHD) was suspected and replacement with hydrocortisone acetate (20 mg/m2/day) and fludrocortisone (0.1 mg/day) was initiated. After clinical improvement and normalization of blood tests, 20 days later (Na+: 140 mEq/L; K+: 4.8 mEq/L; 17-OHP: 31 ng/dL; androstenedione: 0.3 ng/mL), treatment was maintained and the child was discharged.
From that moment on, his blood analyses showed normal-to-low androgen and 17-OHP levels. Throughout his childhood, he maintained high ACTH (ACTH: 2,437 to 3,940 pg/mL) and variable basal cortisol levels (cortisol: 0.5 to 8.2 μg/dl). Besides this, he didn't show dehydration or any other unexpected events. At two years of chronological age (CA), the bone age (BA) was 1.5 years (Greulich-Pyle - GP). On his last visit, at 11 years old, he was on steroid replacement (hydrocortisone 14 mg/m2/day plus fludrocortisone 0.05 mg/day), with good adherence to treatment. Physical examination revealed overweight (40.9 kg; SD =0.82), and adequate stature (height: 139.8 cm; SD = 0.3). The body mass index (BMI) was 20.9 kg/m2, above the 90th centile. He had prepubertal testes, at Tanner I stage. There was hyperpigmentation and dry skin in both cervical areas and internal thighs. Again, ACTH levels were high (670 pg/mL), and basal cortisol levels were in normal range (6.6 μg/dL). Plasma electrolytes were normal.
His younger brother was a term newborn, adequate for gestational age (weight: 2.680 g; length: 47 cm), with no history of prenatal complications. Because he needed treatment for urinary tract infection, he remained in the hospital for 45 days. His initial hormonal profile was not available, as the child stayed in an Intensive Care Unit (ICU), where the diagnosis of adrenal insufficiency was suspected, following his brother pattern. He presented hyponatremia, which could be promptly managed with proper electrolyte and fluid support followed by adequate hormonal replacement with hydrocortisone acetate (20 mg/m2/day) and fludrocortisone (0.1 mg/day). During his first year of life, he showed low levels of plasma 17-OHP (maximum: 20 ng/dL), testosterone (maximum: 294 ng/dL), and androstenedione (maximum 0.7 ng/mL). PRA levels (maximum: 2.3 ng/mL/h) were mildly elevated and ACTH levels were very high (38.901 pg/mL).
In spite of adequate treatment, this child presented an episode of adrenal insufficiency at seven months, but no other unexpected events. When he was four years old, he had 14.3 mg/m2/day of hydrocortisone acetate and fludrocortisone (0.05 mg/day). Physical examination revealed overweight (22.5 kg; SD = 1.72), adequate statural height (106.5 cm; SD = 0.34) and BMI of 19.1 kg/m2 (above the 97th centile). His BA was four years (GP). As for his brother, ACTH levels remained very high (1,201 pg/mL); plasma basal cortisol levels were in normal range (12.3 μg/dL) and there were normal electrolyte concentrations.
During treatment, the patients maintained low 17-OHP and plasma androgens, despite very high ACTH levels, so that we searched for mutations in DAX1 gene in order to establish the etiology of AHC.
Molecular analysis
Children's parents were informed about the importance of establishing the molecular etiology of AHC, in order to better accomplish their follow-up. Written informed consent was obtained from parents for the study and publication. Genomic DNA was isolated from peripheral blood leukocytes of the two siblings and their mother. Both exons of DAX1 gene and the intronic flanking sequences were amplified by polymerase chain reaction (PCR) using specific primers and conditions, as described previously (26). The PCR products were pretreated with an enzymatic combination of shrimp alkaline phosphatase and exonuclease I (United States Biochemical Corp, Cleveland, OH) and directly sequenced using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems, Foster City, CA) in an ABI PRISM 310 automatic sequencer.
DAX1/NR0B1A sequence was compared to ENSG 00000169297 sequence.
RESULTS
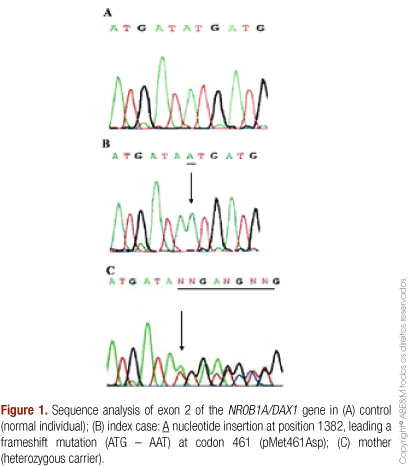
A novel frameshift mutation in exon 2 of DAX1 was identified in the two siblings. An adenine insertion at position 1382 (c1382-1383 insA) causes the substitution of a metionin to an asparagine at codon 461 (ATG → AAT), determining a loss of the stop codon at position 470. The study of mother's DNA confirmed the presence of the same mutation in heterozygous state (Figure 1).
DISCUSSION
Primary adrenal failure is a life-threatening condition that can be caused by a range of etiologies, including autoimmune, metabolic and developmental disorders. DAX1 gene plays an important role in adrenal development and function. Mutations in this transcription factor may cause AHC, a rare cause of primary adrenal insufficiency in childhood (3). Other clinical manifestations may also include isolated mineralocorticoid deficiency (19,27), prolonged testosterone secretion in infancy (28), testicular enlargement (29), genital ambiguity and sex reversal (16,21).
Clinical diagnosis of AHC is not always clearly recognizable and can be overlooked if it is not considered in the differential diagnosis in a context of adrenal crisis. Most of the patients present with failure to thrive, salt-wasting syndrome (hyponatremia, hyperkalemia and metabolic acidosis), hypoglycemia, and skin hyperpigmentation in the first months of life. In some patients, however, adrenal insufficiency may not be evident until they reach adulthood (12).
In the neonatal period, boys generally present signs and symptoms that are indistinguishable from those observed in the salt-losing form of 21-OHD and are frequently misdiagnosed as having this more common disorder (19,29). Moreover, in patients with AHC, plasma steroid determinations may often lead to confusing results in the first weeks of life, probably caused by steroids production by neonates' persisting adrenal fetocortex (29). Distinguishing these two disorders is important because they differ in their clinical course, steroid management and genetic counseling. In the present report, a diagnostic hypothesis of congenital adrenal hyperplasia was firstly made in the elder brother. In the newborn period, the patient presented salt-losing crisis, but very inexpressive elevation of plasma 17-hydroxyprogesterone levels, not similar to the ones currently seen in neonates with the salt-losing form of 21-OHD. Glucocorticoid and mineralocorticoid replacement was initiated with improvement of the children. A diagnosis of AHC was suspected since the patients maintained, throughout their childhood, low 17-OHP and plasma androgens, despite very high ACTH levels and hyperpigmentation. Analysis of the DAX1 gene was essential for the definitive diagnosis of the patients.
Mutational analysis of DAX1 may be worthwhile in any male infant presenting with salt-losing adrenal failure, when steroidogenic disorders, metabolic conditions, syndromes, autoimmune disease, iatrogenic causes, infection and adrenal hemorrhage have been excluded (3).
In the present report, there was no previous family history of adrenal disorders or unexpected death in males. In such families, genetic diagnosis and counseling according to their carrier status should be proposed to all women in reproductive age, to allow earlier treatment of their affected male offspring (1).
Early diagnosis ensures early start of hormonal replacement, diminishing morbidity and preventing sudden death. Moreover, a correct diagnosis has important implications for long-term management and for identifying associated features. Although the hypothalamic-pituitary-gonadal (HPG) axis is often intact in infancy, HH, an associated feature of this disorder, usually becomes apparent during adolescence, with impaired or arrested puberty. These patients will require testosterone replacement for normal development of secondary sexual characteristics. In addition, DAX1 abnormalities may affect testis development and spermatogenesis. Thus, pulsatile GnRH and gonadotropin therapy are often ineffective and the patients may have impaired fertility (23).
In our patients, the molecular analysis confirmed a mutation in exon 2 of DAX1 that generated an abnormal protein. The novel frameshift mutation consisting in adenine insertion at 1382 nucleotide, changing the sequence of amino acids forward at 461 codon and determining a loss of the stop codon at position 470. Probably, this new amino acid sequence in this important region of DAX1 protein prevented its accomplishment in adrenal embryogenesis. The majority of mutations in DAX1 causing AHC/HH reported to date are nonsense or frameshift mutations in the C-terminal region of the gene and have been shown to either impair protein folding and nuclear localization or impair transcriptional repression (5,10,21,30,31).
DAX1 has a unique role as a homologous NR superfamily member, acting as a coregulatory protein that inhibits the transcriptional activity of other NRs (32). Lalli and cols. showed that two domains are necessary for this activity: the N-terminal domain (minimally H3 region) and a C-terminal domain (final portion of H11, H12 regions and the intermediary short loop, including the activation function 2 [AF2] domain) (33). These amino acids of C-terminal domain, which mediates ligand binding, dimerization, and nuclear localization, undergoes allosteric conformational changes in response to ligand binding, through the hormone-dependent transactivation of its AF2 domain (34), located at codons 461-466. Thus, a mutation in this region, which includes the AF2 domain, could affect the domain structure that involved the ligand binding-dependent functions, as dimerization (35).
This novel mutation described here is associated with an early and severe clinical pattern of presentation. As more and more monogenetic disorders are explained on a molecular level, an important question raised is about the correlation of genotype to phenotype. Recent studies highlight the complexities of DAX1 regulation and function (36,37). There is considerable phenotypic variability associated with DAX1 mutations, probably reflecting a combination of genetic (modifier genes, variability in expressivity and penetrance) and environmental (intercurrent illnesses and other stressors) influences (36). To date, genotype-phenotype correlation cannot be easily predicted (38).
In conclusion, we presented a frameshift mutation in the DAX1 gene in two siblings being initially treated as if they had the diagnosis of 21-OHD salt-losing form of CAH, despite the very inexpressive elevation of 17-OHP levels. An insertion of an adenine base (c1382-1383 A ins), which lead to a pMet461Asp substitution, was found. This mutation had not been previously described in the literature. We believe that obtaining a precise genetic diagnosis of this adrenal disorder, which sometimes cannot be confirmed only by clinical aspects, may have important implications for the long-term management of the disease, for predicting prognosis, investigating possible associated features and for appropriate family counseling.
Acknowledgments: the authors thank Sorahia Domenice and Berenice B. Mendonça, from the Unidade de Endocrinologia do Desenvolvimento, Laboratório de Hormônios e Genética Molecular/LIM 42, of the Hospital das Clínicas of the Faculdade de Medicina of Universidade de São Paulo, Brazil, for their technical assistance.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received in Aug/25/2008
Accepted in July/11/2009
- 1. Ostermann S, Salvi R, Lang-Muritano M, Voirol MJ, Puttinger R, Gaillard RC, et al. Importance of genetic diagnosis of DAX1 deficiency: example from a large, multigenerational family. Horm Res. 2006;65(4):163-8.
- 2. Laverty CR, Fortune DW, Beischer NA. Congenital idiopathic adrenal hypoplasia. Obstet Gynecol. 1973;41(5):655-64.
- 3. Lin L, Gu WX, Ozisik G, To WS, Owen CJ, Jameson JL, et al. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (NR5A1) in children and adults with primary adrenal failure: ten years' experience. J Clin Endocrinol Metab. 2006;91(8):3048-54.
- 4. Guo W, Burris TP, Zhang YH, Huang BL, Mason J, Copeland KC, et al. Genomic sequence of the DAX1 gene: an orphan nuclear receptor responsible for X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1996;81(7):2481-6.
- 5. Muscatelli F, Strom TM, Walker AP, Zanaria E, Récan D, Meindl A, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372(6507):672-6.
- 6. Guo W, Burris TP, McCabe ER. Expression of DAX-1, the gene responsible for X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism, in the hypothalamic-pituitary-adrenal/gonadal axis. Biochem Mol Med. 1995;56(1):8-13.
- 7. Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature. 1994;372(6507):635-41.
- 8. Zhang Z, Burch PE, Cooney AJ, Lanz RB, Pereira FA, Wu J, et al. Genomic analysis of the nuclear receptor family: new insights into structure, regulation, and evolution from the rat genome. Genome Res. 2004;14(4):580-90.
- 9. Choi JH, Shin YL, Kim GH, Kim Y, Park S, Park JY, et al. Identification of novel mutations of the DAX-1 gene in patients with X-linked adrenal hypoplasia congenita. Horm Res. 2005;63(4):200-5.
- 10. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. The Human Gene Mutation Database (HGMD®): 2003 Update. Hum Mutat. 2003;21(6):577-81.
- 11. Krone N, Riepe FG, Dörr HG, Morlot M, Rudorff KH, Drop SL, et al. Thirteen novel mutations in the NR0B1 (DAX1) gene as cause of adrenal hypoplasia congenita. Hum Mutat. 2005;25(5):502-3.
- 12. Calliari LE, Longui CA, Rocha MN, Faria CD, Kochi C, Melo MR, et al. A novel mutation in DAX1 gene causing different phenotypes in three siblings with adrenal hypoplasia congenita. Genet Mol Res. 2007;6(2):177-83.
- 13. Mericq V, Ciaccio M, Marino R, Lamoglia JJ, Viterbo G, Rivarola MA, et al. A new DAX-1 mutation in a family with a case of neonatal adrenal insufficiency and a sibling with adrenal hypoplasia and sudden death at 3 years of age. J Pediatr Endocrinol Metab. 2007;20(9):1039-43.
- 14. Yang F, Hanaki K, Kinoshita T, Kawashima Y, Nagaishi JI, Kanzaki S. Late-onset adrenal hypoplasia congenita caused by a novel mutation of the DAX-1 gene. Eur J Pediatr. 2009;168(3):329-31.
- 15. Calvari V, Alpigiani MG, Poggi E, Podesta B, Camerino G, Lorini R. X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism: report on new mutation of the DAX-1 gene in two siblings. J Endocrinol Invest. 2006;29(1):41-7.
- 16. Smyk M, Berg JS, Pursley A, Curtis FK, Fernandez BA, Bien-Willner GA. Male-to-female sex reversal associated with an approximately 250 kb deletion upstream of NR0B1 (DAX1). Hum Genet. 2007;122(1):63-70.
- 17. Ahmad I, Paterson WF, Lin L, Adlard P, Duncan P, Tolmie J, et al. A novel missense mutation in DAX-1 with an unusual presentation of X-linked adrenal hypoplasia congenita. Horm Res. 2007;68(1):32-7.
- 18. Verrijn Stuart AA, Ozisik G, de Vroede MA, Giltay JC, Sinke RJ, Peterson TJ, et al. An amino-terminal DAX1 (NROB1) missense mutation associated with isolated mineralocorticoid deficiency. J Clin Endocrinol Metab. 2007;92(3):755-61.
- 19. Reutens AT, Achermann JC, Ito M, Gu WX, Habiby RL, Donohoue PA, et al. Clinical and functional effects of mutations in the DAX1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab. 1999;84(2):504-11.
- 20. Tabarin A, Achermann JC, Recan D, Bex V, Bertagna X, Christin-Maitre S, et al. A novel mutation in DAX1 causes delayed-onset adrenal insufficiency and incomplete hypogonadotropic hypogonadism. J Clin Invest. 2000;105(3):321-8.
- 21. Seminara SB, Achermann JC, Genel M, Jameson JL, Crowley WF Jr. X-linked adrenal hypoplasia congenita: a mutation in DAX1 expands the phenotypic spectrum in males and females. J Clin Endocrinol Metab. 1999;84(12):4501-9.
- 22. Habiby RL, Boepple P, Nachtigall L, Sluss PM, Crowley Jr WF, Jameson JL. Adrenal hypoplasia congenita with hypogonadotropic hypogonadism: evidence that DAX1 mutations lead to combined hypothalamic and pituitary defects in gonadotropin production. J Clin Invest. 1996;98(4):1055-62.
- 23. Mantovani G, Ozisik G, Achermann JC, Romoli R, Borretta G, Persani L, et al. Hypogonadotropic hypogonadism as a presenting feature of late-onset X-linked adrenal hypoplasia congenita. J Clin Endocrinol Metab. 2002;87(1):44-8.
- 24. Achermann JC, Meeks JJ, Jameson JL. Phenotypic spectrum of mutations in DAX1 and SF-1. Mol Cell Endocrinol. 2001;185(1-2):17-25.
- 25. Achermann JC, Silverman BL, Habiby RL, Jameson JL. Presymptomatic diagnosis of Xlinked adrenal hypoplasia congenita by analysis of DAX1. J Pediatr. 2000;137(6):878-81.
- 26. Yanase T, Takayanagi R, Oba K, Nishi Y, Ohe K, Nawata H. New mutations of DAX1 genes in two Japanese patients with X-linked congenital adrenal hypoplasia and hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1996;81(2):530-5.
- 27. Wiltshire E, Couper J, Rodda C, Jameson JL, Achermann JC. Variable presentation of X-linked adrenal hypoplasia congenita. J Pediatr Endocrinol Metab. 2001;14(8):1093-6.
- 28. Argente J, Ozisik G, Pozo J, Teresa Muñoz M, Soriano-Guillén L, Larry Jameson J. A novel single base deletion at codon 434 (1301delT) of the DAX1 gene associated with prepubertal testis enlargement. Mol Genet Metab. 2003;78(1):79-81.
- 29. Peter M, Viemann M, Partsch CJ, Sippell WG. Congenital adrenal hypoplasia: clinical spectrum, experience with hormonal diagnosis, and report on new point mutations of the DAX1 gene. J Clin Endocrinol Metab. 1998;83(8):2666-74.
- 30. Binder G, Wollmann H, Schwarze CP, Strom TM, Peter M, Ranke MB. X-linked congenital adrenal hypoplasia: new mutations and long-term follow-up in three patients. Clin Endocrinol (Oxf). 2000;53(2):249-55.
- 31. Lehmann SG, Lalli E, Sassone-Corsi P. X-linked adrenal hypoplasia congenita is caused by abnormal nuclear localization of the DAX-1 protein. Proc Natl Acad Sci U S A. 2002;99(12):8225-30.
- 32. Niakan KK, McCabe ER. DAX1 origin, function, and novel role. Mol Genet Metab. 2005;86(1-2):70-83.
- 33. Lalli E, Bardoni B, Zazopoulos E, Wurtz JM, Strom TM, Moras D, et al. A transcriptional silencing domain in DAX-1 whose mutation causes adrenal hypoplasia congenita. Mol Endocrinol. 1997;11(13):1950-60.
- 34. Iyer AK, McCabe ER. Molecular mechanisms of DAX1 action. Mol Genet Metab. 2004;83(1-2):60-73.
- 35. Zhang YH, Guo W, Wagner RL, Huang BL, McCabe L, Vilain E, et al. DAX1 mutations map to putative structural domains in a deduced three-dimensional model. AM J Hum Genet. 1998; 62(4):855-64.
- 36. McCabe ER. DAX1: Increasing complexity in the roles of this novel nuclear receptor. Mol Cell Endocrinol. 2007;265-266:179-82
- 37. Iyer AK, Zhang YH, McCabe ER. Dosage-sensitive sex reversal adrenal hypoplasia congenita critical region on the X chromosome, gene 1 (DAX1) (NR0B1) and small heterodimer partner (SHP) (NR0B2) form homodimers individually, as well as DAX1-SHP heterodimers. Mol Endocrinol. 2006;20(10):2326-42.
- 38. Achermann JC, Ito M, Silverman BL, Habiby RL, Pang S, Rosler A, et al. Missense mutations cluster within the carboxyl-terminal region of DAX1 and impair transcriptional repression. J Clin Endocrinol Metab. 2001;86(7):3171-5.
Publication Dates
-
Publication in this collection
28 Oct 2009 -
Date of issue
Aug 2009
History
-
Accepted
11 July 2009 -
Received
25 Aug 2008