Abstracts
The congenital muscular dystrophies (CMDs) are a group of genetically and clinically heterogeneous hereditary myopathies with preferentially autosomal recessive inheritance, that are characterized by congenital hypotonia, delayed motor development and early onset of progressive muscle weakness associated with dystrophic pattern on muscle biopsy. The clinical course is broadly variable and can comprise the involvement of the brain and eyes. From 1994, a great development in the knowledge of the molecular basis has occurred and the classification of CMDs has to be continuously up dated. We initially present the main clinical and diagnostic data concerning the CMDs related to changes in the complex dystrophin-associated glycoproteins-extracellular matrix: CMD with merosin deficiency (CMD1A), collagen VI related CMDs (Ullrich CMD and Bethlem myopathy), CMDs with abnormal glycosylation of alpha-dystroglycan (Fukuyama CMD, Muscle-eye-brain disease, Walker-Warburg syndrome, CMD1C, CMD1D), and the much rarer CMD with integrin deficiency. Finally, we present other forms of CMDs not related with the dystrophin/glycoproteins/extracellular matrix complex (rigid spine syndrome, CMD1B, CMD with lamin A/C deficiency), and some apparently specific clinical forms not yet associated with a known molecular mechanism. The second part of this review concerning the pathogenesis and therapeutic perspectives of the different subtypes of CMD will be described in a next number.
congenital muscular dystrophy; MDC1A; collagen VI related disorders; glycosylation of alpha-dystroglycan; Fukuyama DMC; muscle-eye-brain (MEB) disease; Walker-Warburg syndrome; rigid spine syndrome
As distrofias musculares congênitas (DMCs) são miopatias hereditárias geralmente, porém não exclusivamente, de herança autossômica recessiva, que apresentam grande heterogeneidade genética e clínica. São caracterizadas por hipotonia muscular congênita, atraso do desenvolvimento motor e fraqueza muscular de início precoce associada a padrão distrófico na biópsia muscular. O quadro clínico, de gravidade variável, pode também incluir anormalidades oculares e do sistema nervoso central. A partir de 1994, os conhecimentos sobre genética e biologia molecular das DMCs progrediram rapidamente, sendo a classificação continuamente atualizada. Nesta revisão apresentaremos os principais aspectos clínicos e diagnósticos dos subtipos mais comuns de DMC associados com alterações do complexo distrofina-glicoproteínas associadas-matriz extracelular que são DMC com deficiência de merosina (DMC tipo 1A), DMCs relacionadas com alterações do colágeno VI (DMC tipo Ullrich e miopatia de Bethlem), DMCs com anormalidades de gliocosilação da alfa-distroglicana (DMC Fukuyama, DMC "Muscle-eye-brain" ou MEB, síndrome de Walker-Warburg, DMC tipo 1C, DMC tipo 1D), além da raríssima DMC com deficiência de integrina. Outras formas mais raras de DMC, não relacionadas com o complexo distrofina-glicoproteínas associadas-matriz extracelular também serão apresentadas (DMC com espinha rígida, DMC tipo 1B, DMC com deficiência de lamina A/C) e, finalmente, algumas formas clínicas com fenótipo aparentemente específico que ainda não estão associadas com um defeito molecular definido. A patogenia e as perspectivas terapêuticas dos principais subtipos de DMC serão apresentados em um próximo número, na segunda parte desta revisão.
distrofia muscular congênita; merosina; colágeno VI; glicosilação da alfa-distroglicana; DMC Fukuyama; DMC "muscle-eye-brain"-MEB; síndrome de Walker-Warburg; espinha rígida
VIEWS AND REVIEWS
Congenital muscular dystrophy. Part I: a review of phenotypical and diagnostic aspects
Distrofia muscular congênita. Parte I: revisão dos aspectos fenotípicos e diagnósticos
Umbertina Conti Reed
Professora Titular da Disciplina de Neurolgia Infantil, Departamento de Neurologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo SP, Brazil
ABSTRACT
The congenital muscular dystrophies (CMDs) are a group of genetically and clinically heterogeneous hereditary myopathies with preferentially autosomal recessive inheritance, that are characterized by congenital hypotonia, delayed motor development and early onset of progressive muscle weakness associated with dystrophic pattern on muscle biopsy. The clinical course is broadly variable and can comprise the involvement of the brain and eyes. From 1994, a great development in the knowledge of the molecular basis has occurred and the classification of CMDs has to be continuously up dated. We initially present the main clinical and diagnostic data concerning the CMDs related to changes in the complex dystrophin-associated glycoproteins-extracellular matrix: CMD with merosin deficiency (CMD1A), collagen VI related CMDs (Ullrich CMD and Bethlem myopathy), CMDs with abnormal glycosylation of alpha-dystroglycan (Fukuyama CMD, Muscle-eye-brain disease, Walker-Warburg syndrome, CMD1C, CMD1D), and the much rarer CMD with integrin deficiency. Finally, we present other forms of CMDs not related with the dystrophin/glycoproteins/extracellular matrix complex (rigid spine syndrome, CMD1B, CMD with lamin A/C deficiency), and some apparently specific clinical forms not yet associated with a known molecular mechanism. The second part of this review concerning the pathogenesis and therapeutic perspectives of the different subtypes of CMD will be described in a next number.
Key words: congenital muscular dystrophy, MDC1A, collagen VI related disorders, glycosylation of alpha-dystroglycan, Fukuyama DMC, muscle-eye-brain (MEB) disease, Walker-Warburg syndrome, rigid spine syndrome.
RESUMO
As distrofias musculares congênitas (DMCs) são miopatias hereditárias geralmente, porém não exclusivamente, de herança autossômica recessiva, que apresentam grande heterogeneidade genética e clínica. São caracterizadas por hipotonia muscular congênita, atraso do desenvolvimento motor e fraqueza muscular de início precoce associada a padrão distrófico na biópsia muscular. O quadro clínico, de gravidade variável, pode também incluir anormalidades oculares e do sistema nervoso central. A partir de 1994, os conhecimentos sobre genética e biologia molecular das DMCs progrediram rapidamente, sendo a classificação continuamente atualizada. Nesta revisão apresentaremos os principais aspectos clínicos e diagnósticos dos subtipos mais comuns de DMC associados com alterações do complexo distrofina-glicoproteínas associadas-matriz extracelular que são DMC com deficiência de merosina (DMC tipo 1A), DMCs relacionadas com alterações do colágeno VI (DMC tipo Ullrich e miopatia de Bethlem), DMCs com anormalidades de gliocosilação da alfa-distroglicana (DMC Fukuyama, DMC "Muscle-eye-brain" ou MEB, síndrome de Walker-Warburg, DMC tipo 1C, DMC tipo 1D), além da raríssima DMC com deficiência de integrina. Outras formas mais raras de DMC, não relacionadas com o complexo distrofina-glicoproteínas associadas-matriz extracelular também serão apresentadas (DMC com espinha rígida, DMC tipo 1B, DMC com deficiência de lamina A/C) e, finalmente, algumas formas clínicas com fenótipo aparentemente específico que ainda não estão associadas com um defeito molecular definido. A patogenia e as perspectivas terapêuticas dos principais subtipos de DMC serão apresentados em um próximo número, na segunda parte desta revisão.
Palavras-chave: distrofia muscular congênita, merosina, colágeno VI, glicosilação da alfa-distroglicana, DMC Fukuyama, DMC "muscle-eye-brain"-MEB, síndrome de Walker-Warburg, espinha rígida.
The congenital muscular dystrophies (CMDs) are genetically and clinically heterogeneous hereditary myopathies with a predominant autosomal recessive mode of inheritance that are characterized by congenital hypotonia, delayed motor development and early onset of progressive muscle weakness, associated with dystrophic pattern on muscle biopsy1,2. The dystrophic pattern is not specific and shared by any type of hereditary muscular dystrophy although it can be variable concerning the amount of histopathological changes. These changes are characterized by great variability in the size of muscle fibers, marked endomysial and perimysial proliferation, and later increase of adipose tissue. In addition, internal nuclei and necrotic as well as regenerating fibers can occur in early stages. The clinical course is broadly variable and can comprise the involvement of the brain and eyes.
The epidemiology of CMD is not well known. Data from an epidemiological study in North-East Italy, considering the period 1979-1993, reported an incidence rate of 4.65 × 10(5) and a prevalence rate of 6.8 × 10(6)3. In Western Sweden, another study considering a similar period (19791994) found a birth incidence of 2.6 × 10(5) and a prevalence of 2.5 × 10(5)4.
HISTORICAL REMARKS
CMD was firstly recognized by Batten who in 19035 and 19046 described the clinical and pathological features of a severe form of congenital myopathy, while the term "Dystrophia Muscularis Congenita" was proposed by Howard7 in 1908. Until the beginning of the last decade of the last century, the CMDs were a highly heterogeneous group of muscular dystrophies of very early onset, whose distinction from other congenital muscular disorders was not clear8. However, a number of distinct forms gradually emerged from the confusing literature on CMD, based on particular clinical or ethnic characteristics8-10. Therefore, the last century was the clinical era of CMD, in which some typical clinical pictures that currently are specific phenotypes were described10:
In 1930, Ullrich11 described a distinct type of CMD named congenital atonic-sclerotic muscular dystrophy, that was characterized by the combination of distal joint hyperelaxity with proximal contractures, scoliosis and severe course;
In 1960, Fukuyama et al.12, described a typical form of severe CMD that was prevalent in Japan and was characterized by the combination of muscular and cerebral involvement; this form was soon recognized as Fukuyama CMD (FCMD);
In 1971, Dubowitz13 described the clinical features of "rigid spine syndrome" that was characterized by an early difficulty in flexing the spine, marked muscular atrophy and joint contractures, as well as progressive scoliosis;
In 1977, Santavuori14 recognized a new form of CMD with cerebral and ocular involvement that named "muscle-eye-brain (MEB) disease"; it was apparently restricted to the Finnish population.
In 1986 the Walker-Warburg syndrome (WWS), whose characteristic brain and eye anomalies had already been described by Walker15 and Warburg16, was definitively classified as a severe form of CMD by Dobyns17 et al., who specified its diagnostic criteria.
At the beginning of the nineties, apart from the cerebro-ocular CMDs, i.e. FCMD, MEB and WWS, the forms of CMDs without central nervous system (CNS) involvement, in general named "pure" or "classical" CMD, were considered a miscellaneous group regarding the severity of the clinical course. From 1990 to 1993, another apparently specific form of CMD emerged from the literature and was characterized by the association of severe muscular involvement and white matter brain changes on neuroimaging in children with normal or near normal intelligence18-25. This form was named "occidental type of cerebromuscular dystrophy"24 for differentiating it from FCMD that was very common in Japan.
In 1993, the molecular era of the CMDs had its onset. Toda et al.26 assigned the gene for FCMD to 9q and soon after, in 1994, Tomé et al.27 by means of immunohistochemical analysis on tissue samples obtained from muscular biopsies found that the severe form of occidental CMD with diffuse white matter abnormality was caused by the lack of laminin M (merosin) chain in the extracellular matrix. This specific form of CMD was classified as MDC1A, and was traditionally termed merosin-negative CMD or CMD with merosin deficiency. The nomenclature for the laminins was soon after revised, and merosin was renamed as laminin alpha-228. The historical background of the CMDs, the steps that allowed the identification of MDC1A and its particularities were revised by Tomé8.
In 1998, Kobayashi et al.29 identified the responsible gene for FCMD, FKTN gene, and its product, a 461-amino acid protein, which they termed fukutin.
Also in 1998, Moghadaszadeh et al.30 identified a locus on chromosome 1p3536 for the peculiar form of CMD with early rigid spine. This genotype/phenotype correlation was the first that allowed the identification of a specific subtype within the classical merosin-positive CMD group.
At the same time, a new, apparently specific clinical form was reported by Muntoni et al.31 in children from the United Arab Emirates who presented CMD with muscle hypertrophy, and early respiratory failure with severe diaphragmatic involvement. Their muscle biopsy samples showed a deficiency of laminin alpha-2 that was considered a secondary phenomenon, since linkage to the LAMA2 locus on 6q22q23 was excluded.
In 1999, the MEB form of CMD was assigned to 1p32p34 by linkage analysis in eight families from which seven were Finnish but one was Turkish32, therefore demonstrating that the MEB phenotype was not restricted to the Finnish population.
In 2000, a linkage analysis in the family from the United Arab Emirates that had already been described by Muntoni et al.31, identified the locus 1q42 that was also confirmed in a second German family with two affected children showing the same respiratory changes and secondary laminin alpha-2 deficiency33. This new genotype/phenotype correlation was named MDC1B33.
In 2001, the locus 1p35-36 that had been assigned to CMD with rigid spine was refined and associated with the gene SEPN1, which encodes the selenoprotein N34. This study by Moghadaszadeh et al.34 was the first to report a human disease caused by selenoprotein dysfunction.
Again in 2001, the first genotype/phenotype correlation related with Ullrich atonic-sclerotic CMD was defined by Camacho et al.35 who identified mutations in one of the three genes coding for collagen type VI (COL6A2, 21q22.3) in three children with Ullrich phenotype.
In 2002 and 2003 a second and a third genotype/phenotype correlation related with Ullrich atonic-sclerotic CMD were reported: Demir et al.36 found mutations in COL6A3 gene (2q37) and Pan et al.37 in COL6A1 gene (21q22.3), therefore illustrating the wide spectrum of genotype-phenotype correlations associated to collagen VI deficiency.
Finally, since 2001, a new pathogenic key for understanding the severe forms of CMDs, most of them associated with CNS changes, emerged from different works38-44 and new genotype/phenotype correlations were established. In children with inability to walk and muscle hypertrophy but no CNS changes and whose muscle samples showed a decreased expression of alpha-dystroglycan (DG), Brockington et al.38 identified a gene at 19q13.3 that codifies the protein fukutin related (FKRP). They named this new genotype/phenotype association as MDC1C and suggested that the mutations in the FKRP gene causing a defective glycosilation (hypoglycosilation) of alpha-DG would explain the basic pathologic mechanism in MDC1C38. Hayashi et al.39 found that also fukutin, whose gene is mutated in FCMD, was involved in the glycosylation of alpha-DG that, when altered, induces a disruption of the extracellular surface membrane in the muscular fiber, also influencing CNS development39. Yoshida et al.40 found that MEB gene codifies a glycosyltransferase, protein O-manose beta1,2-N-acetylglucosaminyltransferase (POMGnT1), involved in O-mannosyl glycosylation of alpha-DG, and suggested that an altered glycosylation could be a new pathomechanism for CMDs with neuronal migration disorders40. These results identified alpha-DG as having an essential role in both muscle and brain development and function44 and inaugurated a new field of research and a new subgroup of CMDs: alpha-dystroglycanopathies or defects of O-glycosilation of alpha-DG. Soon, also WWS was prooved to be an alpha-dystroglycanopathy, and mutations in the gene that codifies the glycosiltransferase protein O-mannosylglycantransferase 1 (POMT1)45 were found in the disease. In 2003, a new rare form of CMD with severe CNS involvement, MDC1D, caused by mutations in the gene that codifies the glycosiltransferase LARGE, was included in this subgroup46. Finally, in 2005, mutations in the gene that codifies the protein O-mannosylglycantransferase 2 (POMT2) was associated to WWS47.
Concerning our knowledge on CMD, after a long clinical era, the transition to the molecular era was surprisingly quick. In 2004, Muntoni and Voit9 revised the CMDs in a work with the following title: "The congenital muscular dystrophies in 2004: a century of exciting progress". The European Neuromuscular Center have periodically organized in Naarden, The Netherlands international workshops on CMD, the first of them in 1993, before the description of CMD1A18. The growing complexity of the knowledge about CMD during the molecular era has been discussed in many workshops48-52, the last one in 200552. Regarding world literature on CMD, a recent report by Fukuyama53 illustrated the great contribution offered by Japanese authors. This fact occurred as FCMD was the first subtype of CMD that was clinically and genetically identified; in addition FCMD provided the new concept of the possibility of extra-muscular involvement, later highlighted with the recognition of the defects of alpha-DG glycosylation. Fukuyama53 reviewed the literature on CMD from 1993 until 2006 and among works, found 726 written by Japanese authors.
The present review includes molecular, clinical and diagnostic data obtained from the recent world literature concerning the different subtypes of CMD. The summary of the details related to each of the subtypes is available on-line by consultation of OMIM. The pathogenic mechanisms and therapeutic perspectives will be described in the second part of the review, in the next number of this journal.
CLASSIFICATION AND GENERAL CLINICAL DATA
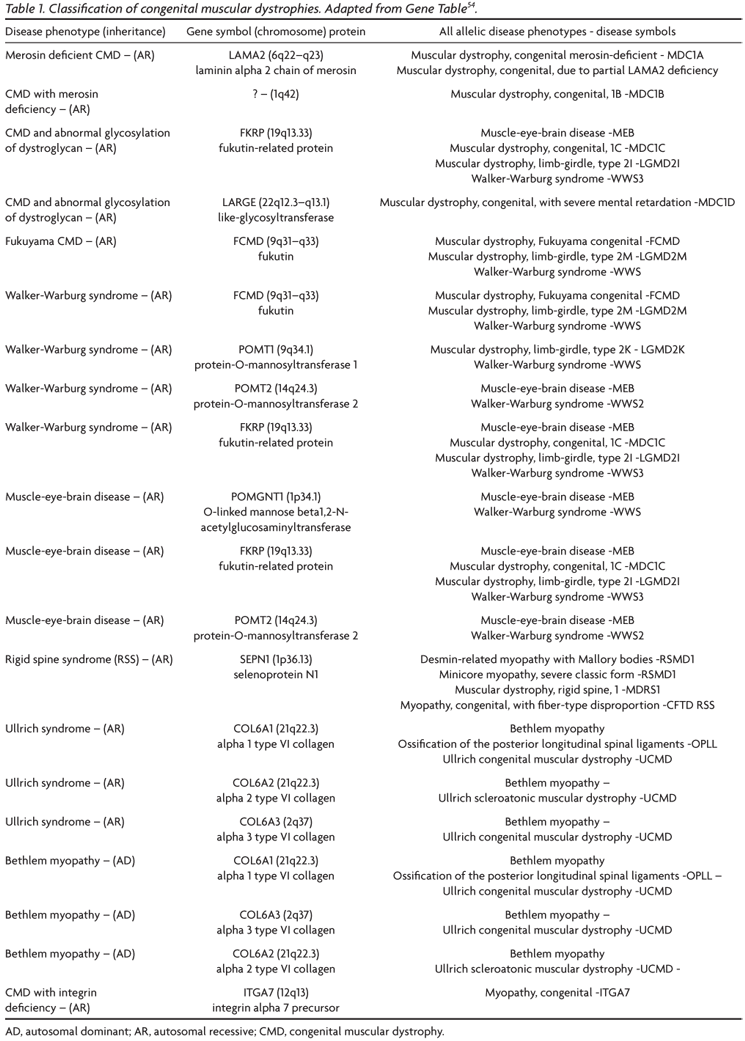
The great development in the knowledge of the molecular basis has allowed a classification of CMDs according to the primary genetic and biochemical defects; this classification has to be continuously up dated. The official journal of the World Muscular Society, Neuromuscular Disorders, periodically publishes the revised classification (Table 1)54. A computerized version of the classification is accessible at http://www.musclegenetable.org and http://194.167.35.195/. Most of the different genes involved with the pathogenesis of the CMDs are related to the function of the dystrophin-glycoproteins associated complex (DAG) in the sarcolemma, either leading to abnormalities of extracellular matrix proteins or of glycosilation of alpha-DG (Fig 1). One of the CMD subtypes is related to a defect of an endoplasmic reticulum protein, selenoprotein N (SEPN1 gene), and recently a new subtype55 was associated to a defect of a nuclear protein, lamin A/C (LMNA gene).
However, as a single gene can be associated to a number of different phenotypes, for the clinician it is more useful to describe the subtypes according to the combination of clinical aspects and primary or secondary protein defect56. A classification of CMDs into three major groups based on the location of the abnormal protein (extracellular matrix, alpha-DG receptors for the extracellular matrix or endoplasmic reticulum) can also be adopted57. In a complete revision on CMDs, Voit and Tomé9 emphasized that the combination of clinical, biochemical and molecular genetic findings must be considered for obtaining the precise diagnosis and providing genetic counseling. They adopted a classification into four groups and considered it open to assimilating future developments: (1) defect of laminin alpha2 primarily affecting the basement membrane: MDC1A; (2) defects due to abnormal glycosylation of alpha-DG: WWS, MEB, FCMD, MDC1B, MDC1C, MDC1D; (3) disorders leading to prominent contractures: rigid spine and Ullrich CMD; (4) primary or secondary alpha 7 integrin deficiency.
Although the frequencies of the different subtypes of CMDs show regional and ethnic variations, the most common are MDC1A and Ullrich CMD. The first corresponds to 30 to 40% of all CMD forms in the European countries9 and also in Brazil58. Ullrich CMD is emerging as the second most common subtype in Europe9,59, in Japan with a rate of 9,4%after FCMD with a rate of approximately 50%60, and in a large Australasian cohort where it reaches 12%61. In Brazil, there is not a precise estimate about the frequency of Ullrich CMD, probably due to the lack of molecular studies that prevents a correct diagnosis in patients with CMD and distal joint laxity. Concerning the alpha-dystroglycanopathies, it is clear that the continuos advances in this field are allowing an increasing number of correct diagnoses and therefore an increasing of estimated frequency9,62. In a large Australasian cohort, among 45 patients whose biopsies were tested with a battery of antibodies, an abnormal immunostaining for glycosilated alpha-DG was found in 11 (25%) and was the most common immunohistochemical abnormality61. However, among those 11 patients only six had a mutation identified in one of five genes codifying glycosyltransferases that were tested (FKRP, POMT1, POMT2, POMGnT1)61. This fact probably indicates the participation of other glycosyltransferases not yet identified in the complex process of alpha-DG glycosylation. The rarest subtypes of CMD are MDC1D, CMD due to primary alpha-7 integrin deficiency, and MDC1B, respectively, with only one46, three63, and six patients31,33, described until the moment.
The degree of muscular and/or CNS involvement is variable within a spectrum from severe "floppy" infant syndrome with feeding and respiratory troubles often early fatal, to moderate motor delay and mild or moderate limb-girdle involvement during childhood compatible with survival into adult life and relatively good quality of life. Due to the enormous clinical heterogeneity of CMDs, the first step for obtaining the correct diagnosis of a determined subtype is a careful analysis of the clinical keys57, such as: CNS involvement (clinically or by neuroimaging); eyes involvement (retina or anterior chamber); degree of clinical severity and progression; type and time of onset of spine deformities and respiratory troubles; distribution of joint contractures and/or joint laxity; presence of muscle hypertrophy, adducted thumbs and skin changes. Recently, a new phenotypical key has benn included as a suggestion of a subtype of CMD related to collagen VI disorders: "sclerotic" or woody consistency on muscle palpation64.
External ophtalmoplegia including ptosis has been exceptionally described in patients whose clinical and histopathologic signs suggest a CMD65-69. In two siblings with ptosis, a possibly specific CMD phenotype associating adducted thumbs, external ophthalmoplegia, mental retardation and cerebellar hypoplasia was suggested67. In others patients with external ophthalmoplegia, the pathogenic mechanism of the myopathic process was supposed to be related to changes of the syntrophin-dystrobrevin subcomplex65,68.
Cardiac involvement has also been scarcely related. Except for the recently recognized LMNA-related CMD55 and for MDD1C linked to FKRP gene38, there are few reports on cardiomyopathy associated to CMD. Mutations in FKRP gene may also cause dilated cardiomyopathy in patients affected by the non congenital LGMD2I form70. In patients with FCMD cardiac involvement may be observed and is particularly frequent in older children71. However, Murakami et al.72 among relatives of four children with FCMD, surprisingly identified six individuals who had the classic compound heterozygous fukutin (gene FKTN) mutation that is constantly observed in FCMD, but showed normal intelligence, no or minimal limb girdle muscle involvement, and severe dilated cardiomyopathy. In one of these patients a cardiac muscle biopsy revealed altered glycosylation of alpha-DG, similar to that observed in FCMD cases. A possible heart involvement has also been reported in patients from cohorts of non specific merosin-positive CMD73,74 or specific MDC1A75,76. Therefore, patients with any form of CMD should be cardiologically investigated, since supportive cardiac therapy can provide optimal disease management and cardiac complications may markedly influence prognosis and outcome77,78.
Muscle MRI, in combination with clinical evaluation, can contribute to determine the best muscle for biopsy and in general has been indicated in inflammatory myopathies for controlling the therapeutic result. However, its value for indicating specific diagnoses and thus selecting the appropriate genetic test has recently been reported79,80, with emphasis to the fact that it is non invasive and easily applied in children, even without sedation80.
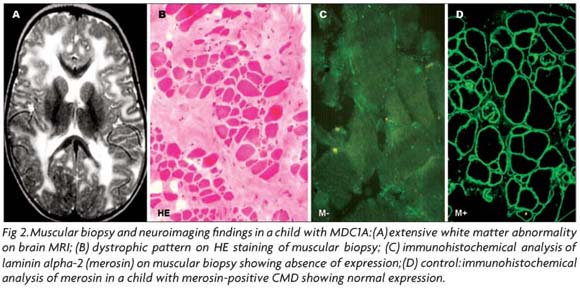
Once a probable phenotype has been selected, as the muscle pathology is usually not specific of any CMD subtype, the immunostaining of muscle biopsy using a battery of antibodies is an essential indicator for the molecular analysis (Fig 2). The immunohistochemical reactions, particularly useful in cases of merosin-deficient CMD, collagen VI-related disorders and CMDs caused by alpha-dystroglycanopathy, will be presented in the topic correspondent to each one of these CMD subtypes.
MDC1A: CONGENITAL MUSCULAR DYSTROPHY WITH ALPHA-2 LAMININ (MEROSIN) DEFICIENCY
MDC1A is caused by mutation in the laminin alpha-2 gene (LAMA2) and was first described by Tomé et al. in 199427. In the same year, Hillaire et al.81 demonstrated that this specific CMD is linked to 6q2, and soon after Heibling-Leclerc et al.82 found the first homozygous mutations in LAMA2 gene in two patients who had been reported by Tomé et al.27. Curiously, according to Di Blasi et al.83, the founder mutation (Cys967Stop) probably originated in Albania.
The clinical spectrum of MDC1A with total absence of laminin alpha-2 chain expression is usually homogeneous corresponding to a severe phenotype characterized by marked muscle weakness and atrophy, diffuse joint contractures, inability to achieve independent ambulation, facial dismorphism, markedly raised creatine kinase (CK) level in blood serum, and characteristic white matter hypodensity on cranial magnetic resonance imaging (MRI)27,58,84-86. Cardiac involvement is rare in patients with MDC1A75-77,87.
Mild allelic variants with partial deficiency of laminin alpha-2 have frequently been reported58,88-93. Generally, these patients present a later onset and slowly progressive weakness that does not avoid the achievement of independent walking, therefore resembling limb-girdle muscular dystrophy (LGMD); they also demonstrate cerebral white matter changes on MRI and peripheral demyelinating neuropathy. It is remarkable that patients with partial reduction of the laminin alpha-2 can have the same severe course that is characteristic of those patients with a total laminin alpha-2 deficiency, and it is impossible to predict the phenotypes based on the amount of protein that is expressed93. However, Tezak et al.94 found that many patients with neonatal-onset and the common severe course have nonsense mutations, while single missense mutations have frequently been reported in milder CMD patients with partial laminin alpha-2 deficiency.
Due to the fact that the alpha-2 subunit of laminin is also expressed in the basal lamina of Schwann cell-axon unit, a peripheral demyelinating neuropathy affecting predominantly motor fibers95, but also the sensitive ones96, is a feature of laminin alpha-2-deficient CMD. However, peripheral motor nerves involvement misses in some patients97 and it has not been found in Brazilian children with MDC 1A58,84, this fact might be due to the type and location of the mutation58.
The characteristic pattern of white matter abnormality associated to MDC1A has been extensively analysed. Opposite to the peripheral nerve, in which laminin alpha 2 is associated only with myelinated and not with unmyelinated nerve fibers and is involved in the myelin stability98, a role for laminin alpha-2 in central myelination has not been confirmed. Villanova et al.99 found that laminin alpha-2 chain is localized to the basal lamina of all cerebral blood vessels and supposed that it may be important for the selective filtration capability of the blood-brain barrier. In patients with MDC1A the lack of laminin alpha-2 may lead to an abnormality of the blood-brain barrier causing impaired selective filtration. Caro et al.100 postulated that disruption of the blood-brain barrier associated with laminin alpha-2 leads to increased water content, resulting in abnormal white matter signal intensity. Using magnetic resonance spectroscopy and apparent diffusion coefficient mapping, Leite et al.101 detected abnormally high free-water concentrations in the white matter of our Brazilian patients with MDC1A and more prominent changes in the parietal, frontal, and temporal white matter. They also found no correlation between the extent of white matter abnormality on MRI and the clinical status as well as the degree of laminin alpha-2 deficiency (partial or total).
Mental subnormality, epileptic manifestations, and neuronal migration defects have been found in few patients102-104. The structural abnormalities mainly involve the occipital cortex103. Recently, Vigliano et al.105 reported a patient with total laminin alpha-2 deficiency and extensive bilateral occipital micropolygyria who presented a mild course during the first six years of life; after this period she started with epileptic seizures and absence-like status and lost ambulation as well as developed cognitive deterioration. She had a homozygous stop-codon mutation in the LAMA2 gene, possibly related with that severe course.
LAMA 2 mutations are markedly variable spanning all protein domains83,94,106 and, in general, the molecular diagnosis is not considered a priority in children with MDC1A, due to the homogeneous phenotype, the easy immunohistochemical analysis of laminin alpha-2 chain in muscle and skin93,107, and the characteristic white matter abnormalities on neuroimaging (Fig 2). However, Siala et al.108,109 have recently emphasized the utility of mRNA analysis in cases of MDC 1A for understanding the mechanism of the mutation and the genotype-phenotype correlation. In addition, the molecular diagnosis is highly recommended for ascertaining the normal status of a second fetus from parents with an afected child, who require genetic counseling. In affected fetuses the immunocytochemical analysis of the trophoblast can detect laminin alpha-2 deficiency; however, DNA samples for linkage analysis to the LAMA2 locus represents the safest method for prenatal diagnosis83,92,110,111.
In patients with the classical MDC1A phenotype in whom the molecular analysis is not essential, the definitive diagnosis is made by muscular biopsy or seldom by skin biopsy as normal skin also expresses laminin alpha2 in the basement membrane at the junction of the dermis and epidermis107. On histopathological evaluation, endo and perimysial fibrosis as well as variation in fiber size, necrosis and adiposis, are more marked in MDC1A when compared with merosin-positive CMD58, 112. In addition, a correlation between the clinical severity and the amount of histopathological changes can be observed in laminin alpha-2-deficient CMD but no in merosin-positive patients112. The immunohistochemical analysis of laminin alpha-2 expression is universally applied and the main particularities about the most efficient antibodies have been reported by Sewry et al.91. The importance of using antibodies directed against different domains of the protein and of refining the epitopes of the commercial antibodies has been emphasized113. He et al.113 reported a patient with partial deficiency of laminin alpha-2, mild nonprogressive muscle weakness and white matter hypodensity, whose muscle biopsy demonstrated an absence of the laminin alpha-2 chain in muscle fibers with two antibodies, but not with four others.
COLLAGEN VI RELATED DISORDERS
Mutations in each one of the three collagen VI genes, COL6A1 (21 q22.3), COL6A2 (21 q22. 3), and COL6A3 (2 q37) that encode respectively the alpha-1, alpha-2 and alpha-3 chains of collagen VI, cause two types of muscle disorders: Bethlem myopathy, with mild or moderate phenotype, and Ullrich CMD, with severe phenotype. Until few years ago, Bethlem myopathy and Ullrich CMD were separate entities with distinct modes of inheritance; presently, the concept that they probably form a spectrum of collagen VI-related disorders with marked clinical and genetic heterogeneity has emerged from the recent advances on the molecular mechanism of both diseases114 and on their complex genotype/phenotype correlations35-37,59,60,114-130.
Bethlem myopathy
Bethlem myopathy is an autosomal dominant inherited disorder caused by mutations in COL6A1, COL6A2 and COL6A359,114,116,118. It was first reported by Bethlem and van Wijngaarden131, in 1976, in three families with 28 affected members, who showed a benign and slowly progressive myopathy. The onset may be in the neonatal period, childhood or adolescence and early contractures of the interphalangeal joints of the fingers, elbows and ankles joints represent a hallmark of this phenotype59,114,118,131,132. Bethlem myopathy is clinically heterogeneous and although in general its clinical course is thought to be benign, Jobsis et al.132, following-up 23 children and 36 adults, found that nearly all children exhibited weakness or contractures during the first two years of life. They132 emphasized that Bethlem myopathy can be slowly progressive and culminate in wheelchair use. As in others collagen-related disorders, follicular hyperkeratosis and keloid formation may be observed in patients with Bethlem myopathy114.
Histopathological findings on muscle biopsy were either nonspecific or compatible with dystrophic changes, and CK levels can be normal or mildly elevated114. Although the dystrophic pattern had previously been considered rare131 or non compatible with Bethlem myopathy, dystrophic changes on the muscular biopsy occur as frequently as the non specific changes116. Muscle immunohistochemistry with Col VI antibodies can be normal in the muscle, and detected only in fibroblast culture that is not a routine procedure114. New immunohistochemical techniques have been tested in an attempt of simplifying the diagnosis of Bethlem myopathy. Recently, Hicks et al.118 applied techniques of immunofluorescent labeling for collagen VI in muscular basal lamina and in fibroblast cultures (skin biopsy-derived)-from 40 patients with genetically confirmed Bethlem myopathy and found that only the fibroblast culture offers conclusive results for the diagnosis of Bethlem myopathy (78%). They also demonstrated that concerning the collagen related disorders the fibroblast immunofluorescent technique is an excellent way for predicting the presence of a COL6A mutation, with a positive predictive value of 75%, a sensitivity and negative predictive value of 100%, and a specificity of 63%. They concluded that the immunofluorescent labeling of collagen VI in fibroblast cultures is a useful diagnostic tool to guide molecular genetic testing in neuromuscular centers that evaluate collagen VI related disorders118. Therefore, in sporadic patients with clinical and histopathological findings suggestive of merosin-positive CMD, who also manifest joint hyperlaxity, the differential diagnosis must include Bethlem myopathy and a fibroblast culture obtained from a simple skin biopsy is recommended.
The data from molecular analysis in Bethlem myopathy revealed that COL6A1 is the most involved gene and a splice site mutation seems to be the most common, not only in COL6A1 as also in COL6A2 and COL6A3 genes116. The skipping of exon 14 in the alpha1(VI) chain has been considered the most common type of mutation in Bethlem patients37,116,124,130. As different kinds of mutations may occur, it has attempted to define genotype/phenotype correlation based on the effect of the mutation on collagen VI biosynthesis, secretion, structure, assembly, and function. Some data indicate that large deletions and mutations inside the triple-helical collagen VI monomer helix formed by alpha-1, 2 and 3 polypeptides are associated with a phenotype more severe than those mutations occurring in the amino-terminal globular region59. Recently, in one patient with Bethlem myopathy it was described a novel mutation that apparently did not affect the assembly, and it was suggested that its effect could be influencing collagen VI interactions in the extracellular matrix116.
Ullrich CMD
The first report of Ullrich phenotype occurred in 1930 by Ullrich11 who named it scleroatonic CMD. However, only in the present century such phenotype has been associated to a muscular deficiency of collagen VI caused by different types of recessive and dominantly acting mutations in the three collagen VI genes35-37. UCMD is clinically less heterogeneous than Bethlem myopathy and the majority of patients have the classic severe form, although others with milder involvement have been reported9. The severe clinical course is characterized by neonatal muscle weakness, proximal joint contractures, hyperlaxity of the distal joints, failure to thrive, lack of independent ambulation, and severe respiratory impairment by the end of the first decade of life9,10,35,36,56,57,59,61,62,114,117,127,133. Intelligence is normal. Other clinical features can include rough skin (follicular hyperkeratosis) or 'sand paper' papular rash56,61,114, hyperhidrosis, congenital hip dislocation, torticollis, prominent ears, facial weakness with a high arched palate, and prominent heels. The healing of wounds is defective and commonly results in the formation of cheloids56. Scoliosis also develops early and further facilitates respiratory complications. CK levels can be normal or increased. The muscle biopsy reveals a typical, in general marked, dystrophic pattern. However in early stages the dystrophic pattern may not be detected and type I fiber atrophy and predominance together with a widening of the fiber diameter spectrum is observed134. In the muscular biopsy of one patient with UCMD, Schessl et al. recently identified the formal diagnostic criteria of histopathological fiber type disproportion. The immunohistochemical analysis of collagen VI in muscle biopsies has given conclusive results, i.e. decreased immunolabelling, but the most secure and helpful immunohistochemical analysis of collagen VI occurs in fibroblast cultures obtained from patients' skin biopsy119. In patients with Ullrich phenotye who without identifiable mutations in the collagen VI genes and normal amount of collagen VI in the interstitium, a primary abnormality of other not yet identified protein interacting with collagen VI in the sarcolemma could cause a failure of collagen VI to anchor the basal lamina to the interstitium. According to Okada et al.60 the possibility of mutations affecting the promoter regions or introns, or of overlooked mutations must be considered in such cases. In Japanese population Okada et al.60 found that mutations in collagen VI genes lead more frequently to a collagen VI deficiency in the sarcollema than in the intersitium. They sequenced the three collagen VI genes in 26 Japanese patients with primary collagen VI deficiency that in Japan accounts for 7.2% of CMD cases. By immunohistochemical analysis they found that most patients had sarcolemma-specific collagen VI deficiency and five had complete collagen VI deficiency, i.e. sarcollema plus interstitium. In the former group all mutations were sporadic dominant; however, in spite of the occurrence of this apparently specific type of mutated collagen VI localization, they could not define any genotype/phenotype correlation.
Initially, molecular studies considered that collagen VI involvement was associated to mutations in collagen VI genes in near to 40% of the patients with Ullrich CMD127. Until 2002 only recessive mutations had been described35,36,59. In 2003, the first heterozygous in-frame deletions acting in a dominantly-negative way was found in the COL6A1 gene37 of one Brazilian patient with severe Ullrich phenotype133; soon, more patients with a dominantly acting mutation in the COL6A1 were reported115 and this same type of mutation has also been found in Ullrich patients with mutations in COL6A2 and COL6A3115. The relatively clinical heterogeneity of Ullrich phenotype, that has been reported9,36,59,117,127 is not related to each of the three loci, but can be associated to the degree of the deficiency of collagen VI in muscle or cultured fibroblasts117. A complete deficiency has been observed in the severe cases while the milder ones that achieve independent ambulation may show a partial deficiency9,117. In general, the complete absence of collagen VI in the extracellular matrix is derived from mutations that exert a strong dominant-negative effect and compromise intracellular assembly of dimers, tetramers, and extracellular microfibrils115. According to Baker et al.115 the new genetic data in Ullrich CMD pointing to the possibility of dominant inheritance37,115,129 highlighted the necessity of a careful molecular investigation for providing an accurate genetic counseling advice.
The definition of genotype/phenotype correlations in collagen VI related disorders is a difficult task due to the number of different mutations and the clinical variability.
In 79 patients with Ullrich CMD or Bethlem miopathy, Lampe et al.123 developed a practical method for analyzing all the 107 exons of the three COL6 genes, achieving over 97% of coverage. This method allowed the identification of mutations in 62% of patients, whose inheritance could be autosomal dominant or recessive. As it had been previously reported37,115 they also identified dominant mutations in a proportion of patients with Ullrich CMD. In addition, several of them had putative recessive mutations each one in a different COL6A gene, a finding that could indicate a novel mode of disease causation or modification. They concluded that mutations are likely to be found in the majority of patients with a clinical diagnosis of Ullrich CMD and Bethlem miopathy; however, the highly polymorphic nature of the three genes suggests the need of specific methods of mutations analysis for performing an adequate genetic counseling123.
Finally, Lampe et al.124 recently compared the molecular data of patients with Ullrich CMD with de novo dominant negative heterozygous splice mutations in COL6A1, COL6A2, and COL6A3, Ullrich CMD with recessively acting splice mutations, and Bethlem myopathy with heterozygous splice mutations. They concluded that in collagen VI related disorders the type of exon skipping mutation dictates the ability of mutant chains for being included in the final multimeric structure of collagen VI microfibrillar network124, and therefore is predictive for clinical severity and inheritance.
Since both Ullrich and Bethlem phenotypes show clinical and genetic heterogeneity, the molecular diagnosis is helpful for the defining the prognosis and for an accurate genetic counseling.
Even considering the possibility of both dominant and recessive mutations in patients with severe Ullrich, a safe genetic counseling is not easy. Peat et al.125, analysing two families with Bethlem and Ullrich phenotype, respectively, found a similar heterozygous mutation causing COL6A1 premature stop codon in the healthy parents of a patient with severe Ullrich phenotype and in heterozygous patients with Bethlem myopathy. In a subsequent pregnancy the parents of the patient with Ullrich phenotype required a prenatal diagnosis and the molecular analysis of the chorionic villus revealed that the fetus was heterozygous for the mutation, therefore eliminating the diagnosis of Ullrich CMD. However, considering that the same type of heterozygous mutation had previously been identified in patients with Bethlem myopathy, the possibility of a mild phenotype could not be excluded in the fetus. The fact that an older proband's brother who was heterozygous showed very mild clinical features reinforced the supposition that the two collagen VI disorders, Bethlem myopathy, and UCMD, belong to a spectrum of collagen VI disorders and are not two separate entities. The authors concluded that in families with homozygous or compound heterozygous null mutations in COL6A1 and probably also in COL6A2 and COL6A3 the genetic counseling deserves a cautious approach as those types of mutation exhibit variable penetrance125.
In conclusion, the different types of inheritance and the great number of possible mutations clearly influence the structure, biosynthesis, secretion, assembly, and functional role of the three collagen VI chains in the extracellular matrix. Ullrich CMD and Bethlem myopathy probably constitute an overlap between the clinical phenotypes and the molecular defects. An overlap among Ullrich CMD, Bethlem myopathy and Ehlers-Danlos syndromes has also been investigated9,59,135. Finally, very recently Merlini et al.64, described two siblings who manifested myosclerosis myopathy associated with mutations in COLA2 gene. The phenotype includes thin muscles with "sclerotic" or woody consistence on palpation and diffuse restriction of movements of all joints, including the jaws. This leads to severe limitation in dayly activities, in spite of a muscle strength relatively conserved. Muscle biopsy showed a partial collagen VI deficiency at the myofiber basement membrane; however, collagen VI was absent around most endomysial/perimysial capillaries. The combination of basement membrane thickening and abnormal pericyte proliferation is suggestive of this rare condition64.
CONGENITAL MUSCULAR DYSTROPHIES CAUSED BY DEFECTS IN THE GLYCOSYLATION OF ALPHA-DYSTROGLYCAN
The defects of the glycosylation of alpha-DG depend on mutations in at least six genes that codify specific or putative glycosyltransferases: POMT145; POMT247; POMGnT40; fukutin29; FKRP38; and LARGE46. Fukutin, FKRP and LARGE are putative glycosyltransferase as until now their exact function and their relation with the known glycosyltransferases, POMT1, POMT2 and POMGnT1, have not been completely elucidated. Six subtypes of CMD and four subtypes of LGMD are caused by deficiency of glycosyltransferase that leads to a hypoglycosylation of alpha-DG. These muscular dystrophies are autosomal recessive disorders, clinically and genetically heterogeneous, that show either pure muscular involvement or different degrees of CNS and/or ophthalmic involvement. The name alpha-dystroglycanopathy is shorter and therefore more pratical and has been commonly utilized although these conditions are not primary defects of alpha-DG. In fact, a primary dystroglycanopathy should be dependent on mutations of the DG gene itself and this possibility has not been identified yet.
The genotypephenotype correlations in muscular dystrophies with defective glycosylation of alpha-DG form a broad clinical spectrum9,10,52,56,57,62,136-139. Within this spectrum, WWS, MEB disease and FCMD belong to the most severe conditions and present different degrees of defective brain migration and eyes abnormalities. On the other side of the spectrum, there are adult patients with LGMD and a pure muscle involvement that can be absolutely variable140. Between these extremes there are other clinical forms of CMD, with or without ocular and CNS involvement, and other LGMD, among which one, LGMD2K141, exceptionally courses with microcephaly and mental retardation.
Although initially these different phenotypes seemed to be associated to mutations in specific genes, the continuous advances and the generalization of the methods for performing molecular diagnosis have increased the number of genotype/phenotype correlations included in the spectrum. Presently, it is accepted that alpha-dystroglycanopathies are overlapping clinical entities and we can summarize their enormous clinical and genetic heterogeneity in Table 2.
FCMD in the Japanese population and MEB in the Finnish population are both due to a founder mutation, retrotransposal29 and splice site142, respectively, and show high regional prevalence. In Japan, FCMD has an incidence around 1 per 10000 births143. In Finland MEB disease shows an average prevalence of 1:50000142. For the other CMDs caused by defects of glycosylation of alpha-DG it is difficult to ascertain prevalence rates. Even for WWS that shows a worldwide distribution, the incidence rate is unknown and has been estimated around 1.2 per 100000 live births3,144.
Clinical manifestations and phenotypic heterogeneity
Voit and Tomé10 reported that within the large spectrum of clinical manifestations in CMDs with glycosylation defects, between the pure muscular involvement and the severe WW phenotype, it is possible to note a hierarchic increase of clinical and radiological severity. They proposed that at one end of the spectrum a mild alteration of alpha-DG glycosylation can cause only myopathic changes and at the other end of the spectrum a marked glycosylation defect results in the severe CNS involvement that accompanies lissencephaly type II. Between the two extremes while the biochemical defect increases there is a progressive worsening of the clinical severity and intermediate but successive stages of variable involvement in cerebellum, pons and eyes10.
The most striking clinical heterogeneity is related to mutations in the FKRP gene. Patients with mutations in the FKRP gene have a broadly variable clinical severity and course: the most severe involvement is represented by prenatal WWS with cobblestone lissencephaly and eye abnormalities defects, and the mildest occurs in subtypes of LGMD with onset in adult life and pure muscular involvement9,10,145. However, approximately 75% of these patients have a LGMD2I phenotype137. Concerning only the FKRP mutations that originate CMD phenotypes, the first description38 had mainly emphasized the phenotype MDC 1C without CNS changes. However, brain involvement is a more common feature than originally anticipated in patients with MDC1C145,146. Mercuri et al.145 described eight patients with FKRP mutations and CNS involvement that was represented by: isolated cerebellar cysts and mental retardation without any other sign of posterior fossa or supratentorial abnormalities in three, and cerebellar cysts associated with structural brain changes involving the posterior fossa and the cortex in five. Among the latter, two patients showed severe brain changes and resembled MEB and WWS-like conditions, respectively, but the other three had various degrees of structural brain abnormalities, including from focal unilateral periventricular nodular heterotopia to marked cerebellar dysplasia and pons hypoplasia. White matter changes were observed in four patients. The authors145 commented that in spite of the fact that in general the severity and the distribution of the white matter changes in patients with FKRP mutations are different from those observed in patients with merosin-deficient CMD, two patients from their cohort had a pattern of white matter abnormality, similar to that of MDC1A. Similarly to Voit and Tomé10, they also suggested that patients with FKRP mutations can show a hierarchy of severity of CNS changes with the cerebellum seeming the most vulnerable structure being the only CNS structure affected in some patients with normal intelligence. In others patients with mental retardation, an increasing clinical and radiological severity can be observed: more extensive cerebellar dysplasias can be accompanied by pons and brainstem abnormalities. Finally, there are patients whose clinical severity is still greater and their MRI reveals structural cortical changes apparently following an anteroposterior gradient. In this study the distribution of FKRP gene mutations did not allow any particular genotype/phenotype correlation concerning the amount of CNS malformations145. However, Keramaris et al.147 considered that the wide phenotypical variation of the FKRP-related myopathies could in part be explained by the type of missense point mutation.
Even considering that mutations in POMT1 and POMT2 are not associated to the same clinical heterogeneity that is observed in the FKRP ones, new mutations in those genes that are not associated to classical WW or MEB phenotypes have been described47,141,148-152. In these occasional case reports many different types and degrees of cortical and posterior fossa malformations have been described and eyes involvement is or is not present; most patients have mental retardation, and calf as well as thigh enlargement has also been related148,149. Yanagisawa et al.150 hypothesized that patients with POMT1 and POMT2 mutations could share the same phenotype because, according to Manya et al.153, both glycosyltransferases form a heterodimeric complex that is responsible for the catalysis of the first step in O-mannosyl glycan synthesis. Recently, Messina et al.154 analysed the genotype of 61 CMD Italian patients with clinical or immunohistochemical pattern suggestive of known forms of alpha-dystroglycanopathies. They found mutations in POMT 1 and POMT2 genes in 30% of the patients with a clear majority in POMT1, 13 cases, than in POMT2, five cases. Mutations causing frameshifts and stop codons were associated to the most severe phenotypes. Only three had a MEB phenotype (POMT1mutation in two and POMT2 mutation in one) while a WWS phenotype was only found in a case with mutations in POMT1. In the remaining 10 patients with POMT1 mutations, six had mental retardation and microcephaly, but normal brain MRI. Predominant cerebellar hypoplasia was common in patients with POMT2 (three out of five) mutations. This study emphasized that like it is observed for FKRP mutations, POMT1 and POMT2 also have a wide clinical and genetic spectrum that is wider than initially thought154.
In 2007, Godfrey et al.137 published the most complete analysis on the genotype/phenotype correlation of alpha-dystroglycanopathies, except those linked to mutations in FKRP gene, in European (including Turkish) and Australian patients. They considered that it is difficult to estimate the mutation frequencies of each one and to recognize the proportion of new phenotypes in relation to the core phenotype that had been originally associated to each of those mutated genes. They reviewed 92 patients in whom FKRP mutation had been already excluded, and whose immunolabelling analysis for alpha-DG in muscle biopsies had showed hypoglycosylation. The patients' DNA was screened for mutations in POMT1, POMT2, POMGnT1, fukutin and LARGE and a mutation was found in only one third of their cohort: mutations in POMT2 were the most common and were found in nine patients, six of them with MEB-FCMD phenotype, two with CMD and cerebellar involvement and one with LGMD associated to mental retardation; mutations in POMT1 occurred in eight patients, most of them with CMD (N=3) or LGMD phenotype (N=3) associated to mental retardation, one with MEB-FCMD phenotype and one with WWS; mutations in POMGnT1 were found in seven patients, six of them with WWS and a single case had LGMD phenotype; mutations in fukutin gene were observed in six patients, four of them without CNS involvement and a single case of each, MEB-FCMD phenotype and WWS; finally, a mutation in LARGE was detected in a single patient with WWS. Therefore they demonstrated that although these genes harbour mutations much less frequently than FKRP gene, the mutations are associated with a striking phenotypical heterogeneity, except for the POMGnT1 gene. However, they pointed to the fact that the LGMD patient with POMGnT1mutation (who was later reported by Clement et al.155) has a mild phenotype and a normal intelligence, therefore "dramatically expanding the phenotypes associated with mutations in POMGnT". Godfrey et al. also137 emphasized that POMT1 and POMT2 mutations were commonly associated with CNS involvement even in patients with mild muscular weakness; the majority of patients with mutations in fukutin gene, opposite to those with typical FCMD, had not structural brain involvement; therefore, mutations in the fukutin gene outside Japan are associated to a milder phenotype. Besides the precise description of the genotype/phenotype correlation, this work also emphasized that patients with CMD and alpha-DG hypoglycosilation in whom any mutation of the known glycosyltransferase genes were found to show a phenotypic spectrum similar to that of patients in whom mutations could be identified: WWS in two; MEB/FCMD in 16; CMD with cerebellar involvement in two; CMD with mental retardation in 12; CMD with pure muscular involvement in nine; LGMD with mental retardation in one, and LGMD with pure muscular involvement in 16. Although out of the aim of their work, the authors137 consider that the amount of depleted glycosylated epitope seems to be correlated with phenotypic severity, so that complete absence in immunostaining would be found in the most severe cases. Apart from the fact that FKRP is in general the most affected gene, the differences of the mutation frequency in the other glycosyltransferases genes may be due to regional influence137. In Italian patients Messina et al.154 found a greater proportion than Godfrey et al.137 of POMT1 and POMT2 mutations and much more mutations in POMT1gene than in POMT2.
Similar to that observed in relation to the phenotype, Godfrey et al.137 did not find any valuable difference in the pattern of dystroglycan expression between patients with and without mutations in any of the genes, a fact that strongly suggested the probable existence of more genes involved on the pathway of glycosylation and not yet identified until the moment137. Cases reports and studies on specific cohorts of patients have emphasized the same probability156-158 and other great evidence comes from the molecular studies from patients with WWS. In spite of its worldwide distribution and therefore the good availability of patients' DNA for genomic analysis, only one third to 40% of the cases has been associated to mutations in one of the six genes involved in the O-manosylation pathways137,159-161.
The amount of alpha-DG immunolabelling on muscular biopsy has been the most precise indicator for the molecular screening; however, to correlate clinical course and alpha-dystroglycan labeling is more commonly observed in patients with mutations in POMT1, POMT2 and POMGnT1 than in those with defects in fukutin and FKRP162.
The molecular analysis is the only method for confirming the alpha-dystroglycanopathy subtype. Considering that the molecular testing is not universally available, it is important to report the possibility of a biochemical approach concerning the three glycosyltransferases that have already been identified. In extracts of muscle biopsies from patients POMGnT1 enzyme activity has been determined using commercially available reagents163. Later, a method using EBV-transformed lymphoblasts, cultured fibroblasts or both fibroblasts and lymphoblasts, has been proposed for the evaluation of POMGnT1, consequently providing the recognition of carriers and the diagnosis of patients with MEB disease164. Recently, lymphoblast-based enzymatic assay has been used for detecting low enzymatic activity of POMGNT1, POMT1 or POMT2 in patients with the respective mutations and has been considered a sensitive method. It can point to only one molecular test; it is also useful for researches in the field of pathogenic mechanisms of glycosyl-transferases165.
In conclusion, WWS, MEB disease, FCMD, CMD 1C and CMD ID are overlapping CMDs collectively designated alpha-dystroglycanopathies, in which the common underlying defect is hypoglycosylation of alpha-DG. We briefly present the characteristic aspects of each one.
Fukuyama congenital muscular dystrophy (FCMD)
FCMD was first described in 1960 by Fukuyama et al.12 and represents one of the most common autosomal recessive disorders in the Japanese population. A recent report on 62 Japanese patients with alpha-dystroglycanopathy, found 54 cases of FCMD, two of MEB disease, one each of WWS and CMD 1C and four cases in which mutation in any known genes associated with glycosylation of alpha-DG could be identified157. FCMD is characterized by CMD associated to cortical migration defects (micropolygyria). In 1993, Toda et al.26 recognized the responsible gene on 9q31-33 (FKTN) and later, in 1998, Kobayashi et al.29 identified the FKTN product, fukutin, a putative glycosyltransferase with 461-amino-acids, whose function and its relation with other alpha dystroglycanopathies has not been yet completely elucidated166. A 3 kb-retrotransposal insertion into the 3' untranslated region of this gene constitutes the founder mutation that derives from a single ancestor and occurs in most FCMD patients in a homozygous or heterozygous manner26,29,143,167,168. This founder mutation is observed in the general population with a frequency of one in 88 individuals and is rare outside the Japanese population29. In addition, at the moment only one FCMD patient with a fukutin homozygous mutation other than the founder mutation has been reported: he had severe brain and eye anomalies and was a Turkish child without Japanese ancestry169.
Fukuyama et al. described the first 15 FCMD cases in 196012 and later he and colleagues reported the details of this particular form of CMD53,170,171. Poor fetal movements and birth asphyxia can be the first signs. Patients manifest generalized muscle weakness and hypotonia from early infancy, mental retardation, and seizures. Hypertrophy of the calves, quadriceps and tongue muscles are commonly seen, as well as a dilated cardiomyopathy that becomes symptomatic in the second decade of life. However the clinical manifestations show a variable degree of severity, inclusively among siblings, and a few patients can walk without support, have a lesser degree of cognitive deficiency and may obtain seizure control. Cerebral and cerebellar micropolygyria, fibroglial proliferation of the leptomeninges, hydrocephalus, focal interhemispheric fusion, and hypoplasia of the corticospinal tracts were the brain malformations originally described by Fukuyama12. Obstructive hydrocephalus is rare. On neuroimaging, transient white matter abnormality that tends to decrease with age, variable occipital cobblestone cortex, hypoplasia of the pons and cerebellar vermis as well as cerebellar cysts can also be revealed172. Ocular abnormalities mainly directed to the retina, such as folding, fusion, focal dysplasia, and detachment have been reported in a high number of patients accompanied or not by other ophthalmological alterations such as abnormal eye movements, strabismus, myopia and microphthalmos173.
Yoshioka and Kuroki174 compared sporadic and familial cases from 41 Japanese families and found that in the familial FCMD patients, motor incapacity was more marked. They emphasized that at the most severe end the broad clinical spectrum of FCMD can include a phenotypic overlap with mild Walker-Warburg syndrome and MEB. Others observed this great variability and found that patients with a compound heterozygous mutation (one allele with the founder mutation and the other with point mutation) have a more severe phenotype than the homozygotes patients175-177. Yoshioka et al.178 compared the epileptic manifestations with the analysis of mutations in 35 FCMD patients and found that the heterozygotes usually develop seizures earlier than homozygotes and can manifest a higher degree of intractable seizures. In addition, in the heterozygotes the mutation different from the 3 kb insertion founder mutation, according to its type, could influence the seizure prognosis178.
In Japanese families, prenatal FCMD diagnosis179 is made by means of haplotype analysis using microsatellite markers. Prenatal testing for detecting disease-causing alleles in members of an affected family is also available179.
Muscle-eye-brain (MEB) disease
MEB disease is an autosomal recessive disorder characterized by CMD, structural eye abnormalities, and cortical malformations. MEB was first described by Raitta et al.180 and Santavuori et al.14 in Finland and is associated with mutations in a gene at 1p34-p3232 that codifies POMGnT1, a proved glycosyl transferase40. The congenital eye abnormalities are variable and include severe myopia, glaucoma, optic nerve hypoplasia and retinal hypoplasia180. Concerning the degree of cortical malformation, CNS involvement is also variable within the spectrum pachygyria/polymicrogyria/agyria associated to posterior fossa changes (flat brainstem and cerebellar hypoplasia) and white matter abnormality14,32,181-183. Other common features include hydrocephalus and dysmorphic face (short nasal bridge, micrognathia, and midface hypoplasia)184. As has been observed in relation to FCMD, MEB also shows a clinical variability, concerning either muscular or mental involvement142,182. Some patients are able to walk and develop speech while others are severely affected with profound motor and cognitive delay or even autistic features185,186. At the lesser degree of severity an unusual mild phenotype can be misdiagnosed as pure CMD. At the most severe end of the spectrum a phenotypic overlap with WWS can occur186 and clinical and radiological particularities may be useful in making the differential diagnosis187,188 between the two conditions.
The spectrum of clinical severity is broad even in the same family: typical MEB phenotype can be observed in one sibling and severe WW-like phenotype in anothe189. Epileptic seizures and obstructive hydrocephalus requiring a shunt have been reported182.
All the mutations in POMGnT1 gene result in a complete loss of enzyme activity, but the type of mutation is apparently not related to the variability in clinical severity186. A single patient with a phenotype of MEB disease and a homozygous mutation in the FKRP gene has been reported187. Probably, mutations in FKRP gene, resulting in either MEB or WWS, affect the glycosyltransferase function in a different way187.
For many years the disorder was considered exclusive to the Finnish population but the advances in molecular diagnosis have been followed by the description of new mutations in POMGnT1 associated to MEB in non Finnish patients190,191 or in patients with atypical phenotypes such as preserved vision190. One study191 reported a slight correlation between the location of the mutation and the clinical severity and emphasized the need for considering POMGnT1 mutations not only in non Finnish MEB-like patients as also in WWS or other CMDs patients around the world191.
Zhang et al.163 described a rapid and relatively simple method for determining POMGnT1 enzyme activity in muscular biopsies using commercially available reagents. They conclude that beyond its diagnostic value in typical MEB patients, the clinical and genetic heterogeneity of CMDs associated with brain and eye malformations justify the use of POMGnT1 assay as a screening procedure for MEB in all those patients163.
Walker-Warburg syndrome (WWS)
WWS received its name from Walker15, who described the first patient with lissencephaly, and from Warburg16 who described a patient with congenital retinal anomalies and hydrocephaly, whose parents were first cousins, therefore defining the autosomal recessive mode of inheritance. Pagon et al.192 described the lack of the typical cortical lamination and due to the combination of hydrocephalus, agyria, as well as retinal dysplasia with or without encephalocele, named the syndrome as HARD ± E. The main clinical manifestations depend on the severe involvement of muscle, eye and brain that in general are not compatible with survival beyond 23 years of age. Affected children do not reach any motor or mental milestone and commonly have seizures and need gastric tube feeding. Supportive measures include hydrocephalus shunting or, occasionally, encephalocele surgical correction144. The muscle involvement is represented by a severe CMD and was characterized as a part of the symptomatic triad by Dobyns et al.17,193, who described the complete clinical picture:brain malformations are typically represented by lissencephaly type II, in which a neuronal overmigration causes a cobblestone cortex; in addition, there is obstructive hydrocephalus that is commonly diagnosed intra uterus, neuronal heterotopias, corpus callosum agenesis, fusion of the hemispheres, leptomeningeal glio-mesodermal proliferation, pontocerebellar hypoplasia with fourth ventricle dilatation, and occasionally occipital encephalocele and Dandy-Walker cyst; the congenital ocular anomalies are both anterior and posterior chamber eye malformations and frequently lead to retinal detachment and blindness; microphthalmia, buphthalmus, optic nerves hyploplasia, colobomas and other iris malformation, congenital or infantile glaucoma, cataract, megalocornea and persistent hyperplastic primary humor vitreous can be observed in variable amounts. Males can present genital anomalies: as animal models show a POMT2 transcript that is specific to the testis, it has been suggested that also the gonadal defects are associated with defects in O-mannosylation194. Rarely, facial dysmorphisms and cleft lip or palate, have been reported144.
Although WWS is clinically homogeneous, it is genetically heterogeneous. Mutations have been found in POMT1, POMT2 and less frequently in POMGnT1, FKRP, fukutin and LARGE genes. In 2002 Beltran-Valero de Bernabe et al.45 studied 10 consanguineous families with WWS and found mutations in POMT1gene in 6 of the 30 unrelated patients. Later, the same researchers found in two unrelated affected girls a homozygous mutation in the in the FKRP and in fukutin gene, respectively187,195. Van Reeuwijk et al.47 described in three unrelated patients, one of them with an affected sib, a homozygous mutation in POMT2. They considered that all patients, independently of which gene was mutated, had the same classical WWS phenotype. Later, they identified in two affected sibs from consanguineous parents a homozygous intragenic deletion in the LARGE gene196. Godfrey et al.137 found a great number of patients with POMGnT1 mutations in their cohort and recently Manzini et al.161 reported that mutations in FCMD gene were more common than previously expected in European/American patients, including all Ashkenazi Jewish cases, who carried the same founder mutation. The genetic heterogeneity makes difficult the prenatal diagnosis in isolated fetuses with hydrocephalus detected by ultrasound. The prenatal ultrasound is very precise in demonstrating type II lissencephaly associated to hydrocephalus197. However, a definitive prenatal molecular diagnosis can only be obtained when in a first affected child the mutation was identified. It has been emphasized that at autopsy of affected fetuses demonstrable muscle changes may not be observed, while brain and eye anomalies are clearly demonstrated197.
Congenital muscular dystrophy type 1C (MDC 1C)
MDC 1C was first reported by Brockington et al.38 who described patients from seven different families with a particularly severe CMD without clinical or radiological CNS involvement. These patients never acquired independent ambulation and had a particular calf and thigh hypertrophy. The course was progressive leading to respiratory failure in the second decade of life and several patients had signs of heart involvement. A marked elevation of serum CK was commonly observed. The authors38 found mutations in the FKRP gene and suggested that the new form was probably caused by a defect of glycosylation of alpha-DG as the muscular biopsies showed a decreased alpha-DG immunostaining. A possible association with clinical and radiological CNS involvement, suggested by mental retardation, cerebellar cysts and white matter abnormalities on neuroimaging, has frequently been reported145,146,198. Brockington et al.140 also reported that mutations in FKRP could cause a LGMD with a relatively benign course, and a wide spectrum of clinical severity depending on the age of onset. Patients with an early onset within the first two years of life presented a Duchenne-like progression with muscle hypertrophy in the legs and eventually in the tongue, and lost independent walking during the second decade of life. In other patients with a variable age of onset and course (some patients were still ambulant in the fifth decade of life) the calf and to a lesser extent, thigh, brachioradialis and tongue muscles were hypertrophic, and muscle cramps following exercise were also relatively common. As it occurred in MDC1C patients, serum CK was markedly increased and a variable degree of heart involvement could be observed. The FKRP mutations identified in the LGMD2I families were different from those seen in MDC1C140.
In patients with FKRP mutations, Brown et al.199 observed that the residual expression of alpha-DG detected by immunolabeling and Western blot can be correlated with the clinical severity and with the type of mutation. Patients with MDC 1C who exhibit a severe phenotype are compound heterozygotes (one missense and one nonsense mutation) or have homozygous missense mutations; they show absent or strongly decreased Ð-DG expression. However, within the spectrum of LGMD2I, patients at the severe end of the clinical spectrum (Duchenne-like) tended to show a greater reduction in immunolabeling of alpha-DG than those at the milder end (later onset) who generally show variable immunohistochemical pattern and sometimes present minimal changes. Patients with the more severe Duchenne-like phenotype are compound heterozygotes (a missense mutation frequently found and either another missense or a nonsense mutation) and patients with the milder form of LGMD2I in general are homozygous for the common C826A/Leu279Ile FKRP mutation199.
Congenital muscular dystrophy type 1D (MDC 1D)
Longman et al.46 analyzed the molecular data from 36 patients with CMD associated to either clinical or radiological signs of CNS involvement or who had abnormal alpha-DG immunolabeling in whom it had been excluded linkage to any known human CMD locus. Among 29 families in which linkage to LARGE gene was not excluded, they found in a 17 year-old girl the first compound heterozygous mutation in this gene. This girl had been a floppy infant with onset of weakness months after birth and manifested severe mental retardation. Neuromaging revealed defects of brain migration and white matter abnormality, and the girl had an abnormal electroretinogram. The muscle biopsy showed a severe dystrophic pattern as well as a decreased alpha-dystroglycan immunolabelling and molecular weight46.
Alpha-dystroglycanopathies: boundaries between CMD and LGMD
In 2004, Kirschner and Bönnemann200 pointed to the fact that the continuous progress in the knowledge of the molecular basis of the muscular disorders was blurring the traditional boundaries between CMD and LGMD, as well as between these and other types of myopathies. In fact, the recognition of the alpha-dystroglycanopathies confirms that sometimes CMD and LGMD share either molecular or pathogenic mechanisms.
LGMD2I, that it was already described above, was the first LGMD to be classified as an alpha-dystroglycanopathy. The original description was made by Driss et al.201 in 13 patients from a Tunisian family, who had a variable age of onset and course, and proximal limb muscle weakness affecting predominantly the pelvic girdle. They map the locus to 19q13.3. Since MDC1C and LGMD2I mapped to the same locus, Brockington et al.140 analysed the molecular data from 25 potential LGMD2I families, including some with a severe and early onset phenotype. In 17 families they found mutations in the FKRP gene, therefore demonstrating that CMD 1C and LGMD2I were allelic disorders.
LGMD2K Dincer et al.151 reported seven patients from six consanguineous Turkish families and an eight sporadic patient from England who manifested a slowly progressive LGMD with mild muscle hypertrophy, increased CK level, microcephaly, and mental retardation. The age of onset varied from one to six years of age. Neuroimaging was normal. The muscular biopsy showed dystrophic pattern and a marked decrease of the glycosilated alpha-DG immunostaining. Later, in some of these patients, Balci et al.141 identified a homozygous mutation in the POMT1 gene that until the moment had only been associated to WWS.
LGMD2M Is the first muscular disorder caused by mutations in fukutin gene that is not associated to CNS clinical or radiological involvement. Godfrey et al.202 reported three children from two families, aged 4 to 10 months, with a severe reduction of alpha-DG in skeletal muscle. In two of them the onset of muscle weakness followed a febrile viral illness and in the other a febrile illness at three years of age was associated with a worsening of the motor symptoms. The patients showed hypertrophy of the lower limb muscles, and increased CK level. All had normal intelligence and neuroimaging. Remarkably, on muscular biopsy the dystrophic features were accompanied by mild macrophage infiltration and the three children had a good response to steroids therapy. Godfrey et al.202 initially suggested classify the new disorder as LGMD2L, but other LGMD had just been associated to the letter L and the new form of alpha-dystroglycanopathy became LGMD2M.
LGMD2N Biancheri et al.203 described a girl with normal intelligence and a mild proximal weakness associated to a marked increase of CK level. The muscular biopsy showed dystrophic and inflammatory changes, as well as marked decrease of alpha-DG that leads to the molecular analysis of the genes involved in alpha-DG glycosylation. A POMT2 homozygous missense mutation was found and Biancheri et al.203 proposed to name this mild LGMD phenotype as LGMD2N.
RIGID SPINE MUSCULAR DYSTROPHY
Rigid spine syndrome, is characterized by marked limitation in the flexion of the spine and gradual development of scoliosis leading to reduced respiratory vital capacity and respiratory failure. Joints contractures at the elbows and the ankles have commonly been described. The first case of this particular CMD phenotype was described by Goebel204 in 1980, and after a long period Moghadaszadeh et al.30, in 1998, and Flanigan et al.205, in 2000, reported respectively five patients from consanguineous families and four siblings from a non consanguineous family with characteristic rigid spine CMD. In one of these affected children a muscle biopsy performed early at nine months of age showed only minimal, nonspecific myopathic changes, but the muscular biopsy of his sib, at 14 years of age, revealed severe dystrophic changes. In some of the patients they found linkage to 1p36p35205. In 2001, Moghadaszadeh et al.34 identified the SEPN1 gene and several mutations resulting in rigid spine CMD. Selenoprotein N is an endoplasmic reticulum glycoprotein involved in muscular function, probably in early development and in muscle cell proliferation or regeneration206. Moghadaszadeh et al.34 also suggested that selenoprotein N may be implicated in redox reactions into the cell, protecting it from oxidant damage.
Rigid spine CMD is rare although no precise data can be reported. Among 11 unrelated patients who had clinical and laboratorial features compatible with SEPN1-related myopathies, Tajsharghi et al.207 found only one patient with rigid spine CMD and pathogenic mutation in SEPN1 gene. Peat et al.61 in a cohort of 101 patients with different subtypes of CMD, also found only one patient with rigid spine CMD and SEPN1 mutation.
In spite of the progressive limitation of the neck and trunk flexion, the progressive axial muscles wasting and the marked generalized muscular atrophy, the patients can maintain the independent ambulation for long periods30,34,205,208,209. The contrast between the fairly well-preserved strength of limb muscles and the severe axial weakness predominantly in neck and trunk weakness with marked scoliosis is particularly impressive209. Facial weakness is a common aspect. The prognosis depends on the degree of involvement of the respiratory function. Respiratory insufficiency has to be early managed with intermittent, positive-pressure, mainly nocturnal ventilation support. Although the scoliosis becomes severe by the end of the first decade in the majority of patients, some children can develop a respiratory insufficiency as early as at three years of age209. Mercuri et al.208 reported a frequent pattern of selective muscle involvement that is observed on muscle imaging: a marked involvement of adductors, sartorius and biceps femoris while rectus femoris and gracilis were relatively spared.
In general the muscular biopsy of patients with rigid spine CMD shows increased variability of fiber size, mild or focal increase of endomysial tissue, regenerating fibers and type I fiber predominance. In some patients small zones of Z-line streaming or a marked dystrophic pattern can be observed. Ferreiro et al.209 performed a molecular analysis of SEPN1 gene in patients with the classical subtype of multiminicore disease because their phenotype with predominant axial weakness and progressive scoliosis with early respiratory involvement was similar to that of rigid spine CMD. They found linkage to the same gene in eight families that had the most severe multiminicore phenotype, some of them with spinal rigidity. The screening for SEPN1 gene in the eight families and in 14 patients with sporadic classical multiminicore myopathy revealed homozygous or compound heterozygous mutations, among which three had already been described in patients with RSMD. The muscle biopsies from patients with rigid spine CMD and multiminicore myopathy differed by the lack of dystrophic changes and by the great amount of typical minicore lesions in the latter. Ferreiro et al. due to the homogeneous clinical features of the two myopathies concluded that rigid spine CMD and the most severe form of classic multiminicore disease are different phenotypes within the same disease spectrum209. Therefore this study demonstrated the existence of overlap and consistent boundaries between congenital myopathies and CMDs.
Later, Ferreiro et al.210, motivated by the finding of Mallory body-like inclusions in two cases of genetically documented SEPN1-related myopathies supposed an identity with a subtype of desmin-related myopathies, which also shows Mallory body-like inclusions. In four patients with CMD from a German family whose clinical and histopathological aspects had already been described by Goebel et al.204 and Fidzianka et al.211, respectively, they demonstrated a linkage to the SEPN1 locus (1p36), and subsequently a homozygous SEPN1 deletion. As the clinical findings were indistinguishable from those of other SEPN1-related myopathies they concluded that this CMD with Mallory body-like inclusions could be included in the same category210.
In 2006 the morphological spectrum of SEPN1-related myopathies was again enriched by the description of SEPN1mutations in two sisters with a diagnosis of typical congenital fiber-type disproportion without minicore lesions or dystrophic changes212. The two adult sisters had manifested from infancy a severe and progressive congenital myopathy with scoliosis and involvement of the thoracic respiratory muscles and represented the first genetically identified form of autosomal recessive congenital fiber-type disproportion. The same mutation was also found in three patients from another family, among whom two had CMD and one had only nonspecific myopathy, without the characteristics pathologic criteria for CFTD. All five patients had abnormal oral glucose tolerance tests and showed biochemical abnormalities suggesting insulin resistance; therefore Clarke et al.212 considered that insulin resistance could be a specific sign in some SEPN1-related myopathies. Like it was previously noted in relation to multiminicore myopathy, this study also represents a narrowing of the boundaries between CMD and congenital myopathies.
Recently, Schara et al.213 reported the phenotype and long-term follow-up in 11 patients with SEPN1-related myopathy, who were teenagers. A broad phenotypic variability was observed and apparently there is no correlation between the type of mutation and the severity of the phenotype. Clinically, the children present as floppy infants with marked delay of motor and mental development. The onset was within the first two years of life with muscle hypotonia. The gross motor development, although with delay, was achieved in most of the patients, who maintain the deambulation for a long period. Rigid spine was observed at a mean age of 10 years. All patients develop respiratory impairment and marked muscular atrophy. Intermittently nocturnally ventilation may be necessary at teen age. The clinical severity was variable and early respiratory failure as well as lack of ambulation could be observed in some patients. However, the degree of respiratory involvement was not related with the degree of weakness and muscular atrophy213.
Okamoto et al.214 developed an antibody for evaluate the immunohistochemical pattern of selenoprotein N and verified that it was diffusely distributed in the cytoplasm of the control muscle, but was reduced and irregularly expressed in the cytoplasm of a patient with rigid spine CMD. In two Japanese patients with rigid spine CMD and different types of mutations, these authors214 also analysed the mutational mechanisms in SEPN1 and the way by which they lead to a truncated selenoprotein N.
CONGENITAL MUSCULAR DYSTROPHY 1B (MDC1B)
Muntoni et al.31 and Brockinton et al.33 described a form of CMD characterized by proximal muscle weakness with marked cervical involvement, muscle hypertrophy, and early respiratory failure. In spite of the occurrence of generalized hypotonia and delayed motor milestones during the first year of life, only one child among six from two unrelated families was reported to manifest the first symptoms at birth. Interestingly the two patients from the second family had manifested episodes of rhabdomyolysis. All patients had normal intelligence and a great increase of CK level. The muscular biopsies from five patients showed a marked dystrophic pattern and deficiency of laminin-a2 that was considered a secondary phenomenon, since linkage to the LAMA2 locus on 6q2223 was excluded. Genomewide linkage analysis of the two families assigned the locus responsible for this form of CMD to chromosome 1q42. The secondary reduction in laminin-a2 chain in these families suggested that the primary genetic defect could affect a gene coding for a protein involved in basal lamina assembly. The authors suggested calling this disorder "CMD1B"33.
CONGENITAL MUSCULAR DYSTROPHY WITH DEFICIENCY OF INTEGRIN ALPHA-7
Hayashi et al.63 described two patients with a compound heterozygosity for two splicing mutations in ITGA7 gene and a third patient with a reduction in integrin alpha-7 mRNA with no observed mutation in the correspondent gene. All children had motor delay compatible to a congenital myopathy but the first also manifested mental retardation without brain MRI changes while the other two manifested congenital torticollis. In one child the muscle biopsy showed mild dystrophic changes with variable fiber size and adipose infiltration but in the others two the muscular biopsy was considered compatible only with congenital myopathy. The immunohistochemical analysis showed a total lack of the integrin alpha-7subunit and normal laminin alpha-2 was normal63. This form of CMD is very rare and no other patients have been described from the original report. In addition, an overt dystrophic aspect was not demonstrated in the affected patients. However, as the mice null allele for integrin-alpha-7 gene typically demonstrated clinical and histopathologic aspects of a progressive muscular dystrophy215, the form of CMD with integrin alpha-7deficiency persists within the CMD classification.
CMD CAUSED BY MUTATION IN LMNA GENE
In a review on CMD, Jimenez-Mallebrera et al.56 considered the possibility that in addition to the defects in the group of proteins already known, new pathological pathways could also emerge. In fact this assertion has been recently confirmed when an apparently specific CMD phenotype characterized by marked cervical weakness was found to be associated with mutations in lamin A/C gene55. Lamins are the major proteins of the nuclear envelope and are located in the inner nuclear membrane that determines nuclear shape and size216. Laminopathies are a highly heterogeneous group of disorders caused by mutations in the LMNA gene, which codes for A-type lamins of the nuclear envelope. Mutations in this gene have been associated to a marked phenotypic heterogeneity that also includes non muscular disorders217,218. Regarding myopathic phenotypes, autosomal dominant Emery-Dreifuss, LGMD1B and muscular dystrophy associated with cardiac conduction system defects are the most common conditions. Congenital onset had been previously found in two children: one with severe predominant axial weakness and poor neck control219 and other with prominent weakness of neck extensor muscles220. These two children showed increased CK level but the findings on muscle biopsies were nonspecific.
Quijano-Roy et al.55 reported 15 patients (11 from different centers including the two mentioned above) with de novo heterozygous LMNA mutations and consistent clinical phenotype, defining a distinct and clinically recognizable form of early-onset myopathy. All children manifested a muscle weakness with onset in the first year of life and a remarkable selective axial weakness with wasting of the cervicoaxial muscles causing "dropped head" syndrome phenotype. They distinguished two subgroups of patients: the first, smaller, with severe weakness and minimal or absent motor development, and the second, larger, with dropped-head syndrome following a normal period of motor development. All children manifested initially a rapid and progressive involvement of cervical/axial muscles, and after, a period of slow or no progression. Progressive restrictive respiratory insufficiency occurred early in the first group but even in the second group was evident before the age of eight years. Four children showed cardiac arrhythmias what recommends a careful cardiac surveillance on follow-up. In addition limb involvement was predominantly proximal in upper extremities and distal in lower extremities, and a rigid spine with thoracic lordosis developed early. The serum CK level was universally increased and mild-to-severe dystrophic changes were seen in almost half of the patients, being much more abnormal in deltoid than quadriceps muscles. Scattered atrophic type 1 fibers were common and could be a diagnostic clue. Since mononuclear infiltrates and/or positive inflammatory markers were found in some patients' muscle biopsies, a steroid therapy was occasionally attempted but it does not modify the degree of weakness. The authors discussed in details the broad clinical spectrum of the laminopathies and, concerning their present series, emphasized that among the causes of dropped-head syndrome, other forms of CMDs, particularly SEPN1-related myopathies and congenital myopathies should be considered for making a correct differential diagnosis. Since many patients had novel mutations or shared the same mutation, they also supposed that the specific mutations identified in their cohort might be related to the severe phenotype. In addition they found that patients with other laminopathies who carried known mutations that were also identified in part of the children from their cohort, had in general, but not ever, a most severe phenotype than is typical for Emery-Dreyfuss muscular dystrophy or LGMD1B. They concluded that modifier genes and/or environmental factors may also contribute to the clinical variability. The authors55 suggested that this new entity is a CMD, and named it LMNA-related CMD, or L-CMD.
With the identification of this new entity, the assertion that the traditional boundaries between the categories of CMD and LGMD are blurring200 can again be demonstrated. Until the moment, these boundaries that had to be reviewed essentially covered the proteins linking the sarcolemma to the extracellular matrix221, as that the involvement of nuclear proteins represents a novel approach for CMDs. However, in a recent paper analyzing the main conditions that can be confused with CMD, Klein et al.222 refer to this early and severe laminopathy within a topic named "Other muscular dystrophies with early onset".
CONGENITAL MUSCULAR DYSTROPHIES NOT YET RELATED WITH A SPECIFIC MOLECULAR DEFECT
Despite the continuous advances that have been reported in the last eight years about molecular and pathogenic aspects of the different subtypes of CMD, there are still clinical forms in which the protein defect is unknown. These clinical forms include phenotypical aspects that may be judged sufficiently distinct to suggest a possible specific entity9.
For example, Voit et al.67 described a form of CMD in two siblings who in addition to the classical, clinical and histological changes compatible with CMD, also have adducted thumbs and toe contractures, ptosis, external ophthalmoplegia, mild mental retardation, and mild cerebellar hypoplasia on MRI. In these patients they excluded the most common loci for CMD that had been identified at that time and emphasized that adducted thumbs was a distinct clinical sign that might be included within the spectrum of phenotypic changes in CMD67.
Vondracek et al.69 have also reported an apparently specific variant of CMD in a girl with normal intelligence and congenital muscular involvement associated to progressive external ophtalmoplegia, and white matter changes on neuroimaging; muscle biopsy revealed a dystrophic pattern with a prominent inflammatory infiltrate. As an electron microscopy showed the accumulation of abnormally enlarged mitochondria under the sarcolemma, a study of respiratory chain enzyme activities and a sequencing of the entire mitochondrial genome were made and the results were unremarkable.
Topaloglu et al.223 reported three cases in two families with merosin-positive congenital muscular dystrophy, mild mental retardation, bilateral cataracts and normal cranial MRI. In our Brazilian cohort, we found similar clinical picture in two siblings with severe CMD, cataract, retinitis pigmentosa, microcephaly, mild mental retardation and normal cranial MRI. The course of the disease, apparently static during the first 10 years of life, became progressive during the second decade with loss of deambulation by the age of 13 years. CK level was increased in both children224,225.
Another peculiarity about CMD is the presentation of familial cases in which a novel locus is suggested with basis on linkage analyses. Mahjneh et al.226 described 10 patients with CMD belonging to two generations of a large consanguineous Palestinian family: the children had onset at birth and show a relatively benign but variable clinical course with more marked weakness in the proximal upper limb-girdle and trunk muscles. Intelligence and brain CT were normal. Later, a genome-wide linkage search of the family permitted the identification of a novel locus on 4p16.3227.
In 2005 Tetreault et al.228described a group of 14 FrenchCanadian patients, from 11 families, suffering from a CMD with joint hyperlaxity (CMDH) with clinical overlap with Ullrich CMD. They share with Ullrich CMD the presence of congenital hypotonia, weakness, contractures, distal joint hyperlaxity that can be concomitantly proximal, scoliosis, normal intelligence and frequent delayed motor milestones. However, the clinical aspects were less severe than in Ullrich CMD: they acquired independent walking and do not manifest respiratory failure despite the scoliosis. In muscle the histopathologic changes were not homogeneous but all the available biopsies showed variation in fiber size, central nuclei and increased endomysial connective tissue. In two families the authors excluded mutations in the three genes coding for collagen VI subunits and established linkage of all families to a region on chromosome 3p2321228.
DIFFERENTIAL DIAGNOSIS
In general, at the first evaluation of a child with CMD, clinical, and soon after, histopathologic and immunohistochemical aspects on the muscular biopsy can indicate a specific genetic testing. However, despite the molecular advances, a number of patients with early weakness and dystrophic pattern on muscle biopsy suggesting CMD can later be recognized as a different condition. A recent work highlights the difficulties in reaching, still in early infancy, a final diagnosis for part of the children who have clinical and histopathologic signs that can be classified as CMD222. Klein et al.222 reported that during the last five years among 400 patients initially diagnosed as CMD in a large center of reference for children with neuromuscular disorders, 25 patients were found to have diagnosis other than CMD. The final diagnosis in these 25 patients were the following: early onset laminopathy in four; congenital facioscapulohumeral muscular dystrophy in one; core myopathy with de novo mutation of the muscle Ryanodine Receptor gene in three; congenital spinal muscular atrophy with predominant lower limb involvement in four children (early muscle biopsy had showed marked fatty infiltration and changes that could be interpreted as myopathic); neurogenic arthrogryposis in one; amyoplasia in one; Marinesco Sjoegren syndrome in two; cerebro-oculo-facio-skeletal syndrome that can clinically overlap with Marinesco Sjoegren syndrome in one; mitochondrial depletion in two children whose multisystemic involvement became apparent after the first year of age (muscle biopsy had showed a few fibers negative or pales on cytochrome C oxidase staining); infantile systemic hyalinosis in one; Pompe disease in one child who showed marked muscle and respiratory involvement with no cardiac involvement until two years of age; Ehlers Danlos syndrome in one child who showed marked skin hyperelasticity and presented severe weakness at birth that started to improve at seven months of age; translocation 9;13 in two patients. They considered that Marinesco-Sjoegren syndrome should be particularly reminded in the differential diagnosis of infants with muscle involvement, as like many forms of CMD, includes mental retardation and other signs of CNS involvement222.
PALLIATIVE TREATMENT AND FINAL REMARKS
Despite the broad field of researches on new therapeutic strategies, at present days, CMD has still to be managed by palliative care and the type of supportive treatment mainly depend on the age of the patient and on the severity of clinical manifestations and complications. Due to the severe and almost invariably progressive course of many subtypes of CMD, the palliative care should include an interdisciplinary staff of health professionals who try to maintain the patient's independence, first in relation to independent walking or mobility, and later controlling upright and sitting posture, as long as possible. For adequately dealing with the associated complications, such as joint contractures, scoliosis and, specially, respiratory insufficiency an integrated medical staff must to keep in mind the patients'quality of life. Orthopedic surgery for releasing contractures, spinal surgery and orthotic devices have to been discussed individually for each patient and his family10.
In relation to stretching, splinting and general physiotherapic measures, as well as occupational therapy and other rehabilitation procedures, the literature does not specifically report on CMD, although neuromuscular disorders of children, in general, and Duchenne Muscular Dystrophy as well as Spinal Muscular Atrophy, in particular, have been extensively approached.
The prevention and management of respiratory complications is a highly developed field of palliative care, so that the outcome and quality of life of children with progressive neuromuscular disorders have clearly improved. Vaccination against common respiratory infections and a careful management of antibiotictherapy. Many authors have described the principles of ventilator support for neuromuscular disorders and the importance of periodic and regular assessments of respiratory function, performing spirometry229-236. Mellies et al.232,233 emphasized particularly the role of noninvasive positive pressure ventilation in the management of these patients. Considering the high clinical variability, the follow-up of patients with any subtype of CMD must also include the evaluation and treatment of sleep disordered breathing by means of oximetry periodic polysomnographic records or simpler overnight pulse oximetry measures, and nocturnal ventilator support237.
Children with merosin deficient CMD have progressive feeding difficulties, including delay in the act of eating, that result in failure to thrive; their nutritional status as well as that of children with other severe forms has to be assessed because can be related to an increase of respiratory infections238. Ramelli et al.239 evaluated the indications of gastrostomy placement in paediatric patients with neuromuscular disorders and noted that this procedure leads to a reduction of the frequency of chest infections and has a good impact on feeding difficulties and malnutrition. In addition they report no significant complications and families' satisfaction.
In conclusion, the phenotypic characterization, the immunohistochemical or quantitative determination of certain proteins in muscle biopsy or fibroblast culture, and eventually enzymatic assays are valuable tools for the clinician in order to select the adequate molecular study. The molecular diagnosis is essential for genetic counselling and prenatal diagnosis when required, but due to the clinical heterogeneity of some subtypes of CMD is not always decisive for attesting the long-term course of the diseases. However, the advances in the field of molecular genetics, cell biology and biochemistry have been the source of a better knowledge on the pathogenic mechanisms involved in the CMDs and consequently in the development of effective therapeutic strategies. Unfortunately, the induction of a functional protein by gene modification, cell therapy or pharmacological agents has been a long and difficult task. The effective therapy is coming but employing it in large-scale clinical trials constitutes a challenge that is much greater in undeveloped countries.
REFERENCES
1. Banker BQ, Engel AG. Basic reaction of muscle. Engel AG, Franzini-Armstrong C. Myology: basic and clinical. Edition 3. New York: McGraw-Hill, 2004:691-748.
2. Dubowitz V, Sewry CA. Muscle biopsy: a practical approach. Edition 3. Philadelphia: Elsevier, 2006:349-376.
3. Mostacciuolo ML, Miorin M, Martinello F, Angelini C, Perini P, Trevisan CP. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet 1996;97:277-279.
4. Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord 2000;10:1-9.
5. Batten FE. Three cases of myopathy, infantile type. Brain 1903;26:147-148.
6. Batten FE. Case of myositis fibrosa with pathological examination. Trans Clin Soc Lond 1904;37:12-22.
7. Howard R. A case of congenital defect of the muscular system (Dystrophia muscularis congenita) and its association with congenital talipes equino-varus. Proc R Soc Med 1908;1 Pathol sect:157-166.
8. Tomé FM. The Peter Emil Becker Award lecture 1998. The saga of congenital muscular dystrophy. Neuropediatrics 1999;30:55-65. Neuromuscul Disord 2003;13:207-215.
9. Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14:635-649.
10. Voit T, Tome FM. The congenital muscular dystrophies. Engel AG, Franzini-Armstrong C. Myology - basic and clinical. Edition 3. New York: McGraw-Hill-Medical Publishing Division, 2004:1203-1238.
11. Ullrich O. Kongenitale atonisch-sklerotische Muskeldystrophie, ein weiterer Typus der heredodegeneration Erkrankungen des nueromuskularen Systems. Z Ges Neurol Psychiat 1930;126:171-201.
12. Fukuyama Y, Kawazura M, Haruna H. A peculiar form of congenital progressive muscular dystrophy: report of fifteen cases. Paediat Univ Tokyo 1960;4:5-8.
13. Dubowitz V. Rigid spine syndrome: a muscle syndrome in search of a name. Proc R Soc Med 1973;66:219-220.
14. Santavuori P, Leisti, J, Kruus S. Muscle, eye and brain disease (MEB): a new syndrome. Neuropädiatrie 1977;8(Suppl):S553-S558.
15. Walker AE. Lissencephaly. Arch Neurol Psychiatr 1942:48:13-29.
16. Warburg M. Heterogeneity of congenital retinal nonattachment, falciform folds and retinal dysplasia: a guide to genetic counselling. Hum Hered 1976:26:137-148.
17. Dobyns WB, Pagon RA, Armstrong D, et al. New diagnostic criteria for Walker-Warburg syndrome. Am J Hum Genet 1986;39:A59.
18. Dubowitz V. 22nd ENMC sponsored workshop on congenital muscular dystrophy held in Baarn, The Netherlands, 14-16 May 1993. Neuromuscul Disord 1994;4:75-81.
19. Kanagawa M, Saito F, Kunz S, et al. Congenital muscular dystrophy of a non-Fukuyama type with white matter hyperlucency on CT scan. Brain Dev 1992;14:420-422.
20. Kao KP, Lin KP. Congenital muscular dystrophy of a non-Fukuyama type with white matter hyperlucency on CT scan.Brain Dev 1992;14:420-422.
21. Oliveira AS, Gabbai AA, Kiyomoto BH, Schmidt B. Congenital muscular dystrophy: report of 8 cases with evidence of central nervous system involvement. Rev Paul Med 1990;108:139-141.
22. Oliveira AS, Gabbai AA, Kiyomoto BH, Ferreira Neto A, Schmidt B, Lima JG. Congenital muscular dystrophy: clinical study of 17 patients. Arq Neuropsiquiatr 1991;49:265-271.
23. Tanaka J, Mimaki T, Okada S, Fujimura H. Changes in cerebral white matter in a case of congenital muscular dystrophy (non-Fukuyama type). Neuropediatrics 1990;21:183-186.
24. Topaloglu H, Yalaz K, Kale G, Ergin M. Congenital muscular dystrophy with cerebral involvement: report of a case of "occidental type cerebromuscular dystrophy"? Neuropediatrics 1990;21:53-54.
25. Trevisan CP, Carollo C, Segalla P, Angelini C, Drigo P, Giordano R. Congenital muscular dystrophy: brain alterations in an unselected series of Western patients. J Neurol Neurosurg Psychiatry 1991;54:330-334.
26. Toda T, Kanazawa I, Nakamura Y. Localization of a gene responsible for Fukuyama type congenital muscular dystrophy to chromosome 9q31-33 by linkage analysis. Human Genome Mapping Workshop 1993:A20.
27. Tome FM, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci 1994;317:351-357.
28. Burgeson RE, Chiquet M, Deutzmann R, et al. A new nomenclature for the laminins. Matrix Biol 1994;14:209-211.
29. Kobayashi K, Nakahori Y. Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998;394:388-392.
30. Moghadaszadeh B, Desguerre I, Topaloglu H, et al. Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigidity of the spine, on chromosome 1p35-36. Am J Hum Genet 1998;62:1439-1445.
31. Muntoni F, Taylor J, Sewry CA, Naom I, Dubowitz V. An early onset muscular dystrophy with diaphragmatic involvement, early respiratory failure and secondary alpha2 laminin deficiency unlinked to the LAMA2 locus on 6q22. Eur J Paediatr Neurol 1998;1:19-26.
32. Cormand B, Avela K, Pihko H, et al. Assignment of the muscle-eye-brain disease gene to 1p32-p34 by linkage analysis and homozygosity mapping. Am J Hum Genet 1999;64:126-135.
33. Brockington M, Sewry CA, Herrmann R, et al. Assignment of a form of congenital muscular dystrophy with secondary merosin deficiency to chromosome 1q42. Am J Hum Genet 2000;66:428-435.
34. Moghadaszadeh B, Petit N, Jaillard C, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nature Genet 2001;29:17-18.
35. Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA 2001;98:7516-7521.
36. Demir E, Sabatelli P, Allamand V, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet 2002;70:1446-1458.
37. Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet 2003;73:355-369.
38. Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha-2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001;69:1198-1209.
39. Hayashi YK, Ogawa M, Tagawa K, et al. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001;57:115-121.
40. Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001;1:717-724.
41. Kano H, Kobayashi K, Herrmann R, et al. Deficiency of alpha-dystroglycan in muscle-eye-brain disease. Biochem Biophys Res Commun 2002;291:1283-1286.
42. Michele DE, Barresi R, Kanagawa M, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002;418:417-422.
43. Moore SA, Saito F, Chen J, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002;418: 422-425.
44. Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet 2002;360:1419-1421.
45. Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71:1033-1043.
46. Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Molec Genet 2003;12:2853-2861.
47. van Reeuwijk J, Janssen M, van den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 2005;42:907-912.
48. Dubowitz V. 68th ENMC International Workshop (5th International Workshop): on Congenital Muscular Dystrophy, 9-11 April 1999, Naarden, The Netherlands. Neuromuscul Disord 1999;9:446-454.
49. Muntoni F, Guicheney P. 85th ENMC International Workshop on Congenital Muscular Dystrophy. 6th International CMD Workshop. 1st Workshop of the Myo-Cluster Project 'GENRE'. 27-28th October 2000, Naarden, The Netherlands. Neuromuscul Disord 2002;12:69-78.
50. Muntoni F, Bertini E, Bönnemann C, et al. 98th ENMC International Workshop on Congenital Muscular Dystrophy (CMD), 7th Workshop of the International Consortium on CMD, 2nd Workshop of the MYO CLUSTER project GENRE. 26-28th October, 2001, Naarden, The Netherlands. Neuromuscul Disord 2002;12:889-896.
51. Muntoni F, Valero de Bernabe B, Bittner R, et al. 114th ENMC International Workshop on Congenital Muscular Dystrophy (CMD) 17-19 January 2003, Naarden, The Netherlands: (8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE). Neuromuscul Disord 2003;13:579-588.
52. Muntoni F, Voit T. 133rd ENMC International Workshop on Congenital Muscular Dystrophy (IXth International CMD Workshop) 21-23 January 2005, Naarden, The Netherlands. Neuromuscul Disord 2005;15:794-801.
53. Fukuyama Y. Fukuyama congenital muscular dystrophy-history and perspectives. Brain Nerve 2008;60:43-51.
54. Gene table of monogenic neuromuscular disorders (nuclear genome only). Neuromusc Disord 2008;18:101-129.
55. Quijano-Roy S, Mbieleu B, Bönnemann CG, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008;64:177-186.
56. Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F. Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 2005;62:809-823.
57. Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006;9:427-443.
58. Ferreira LG, Marie SK, Liu EC, et al. Dystrophin-glycoproteins associated in congenital muscular dystrophy: immunohistochemical analysis of 59 Brazilian cases. Arq Neuropsiquiatr 2005;63:791-800.
59. Pepe G, Bertini E, Bonaldo P, et al. Bethlem myopathy (BETHLEM) and Ullrich Scleroatonic Muscular Dystrophy: 100th ENMC International Workshop, 23-24 November 2001, Naarden, The Netherlands. Neuromuscul Disord 2002;12:984-993.
60. Okada M, Kawahara G, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69:1035-1042.
61. Peat RA, Smith JM, Compton AG, et al. The diagnosis and etiology of congenital muscular dystrophy. Neurology 2008;71:312-321.
62. Mercuri E, Longman C. Congenital muscular dystrophy. Pediatr Ann 2005;34:560-562.
63. Hayashi YK, ChouFL, Engvall E, et al. Mutations in the integrin alpha-7 gene cause congenital myopathy. Nature Genet 1998;19:94-97.
64. Merlini L, Martoni E, Grumati P, et al. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 2008;71:1245-1253.
65. Jones KJ, North KN. External ophthalmoplegia in neuromuscular disorders: case report and review of the literature. Neuromuscul Disord 1997;7:143-151.
66. Salih MA, Al Rayess M, Cutshall S, et al. A novel form of familial congenital muscular dystrophy in two adolescents. Neuropediatrics 1998; 29:289-293.
67. Voit T, Parano E, Straub V, et al. Congenital muscular dystrophy with adducted thumbs, ptosis, external ophthalmoplegia, mental retardation and cerebellar hypoplasia: a novel form of CMD. Neuromuscul Disord 2002;12:623-630.
68. Jones KJ, Compton AG, Yang N, et al. Deficiency of the syntrophins and alpha-dystrobrevin in patients with inherited myopathy. Neuromuscul Disord 2003;13:456-467.
69. Vondracek P, Hermanova M, Vodickova K, et al. An unusual case of congenital muscular dystrophy with normal serum CK level, external ophtalmoplegia, and white matter changes on brain MRI. Eur J Paediatr Neurol 2007;11:381-384.
70. D'Amico A, Petrini S, Parisi F, et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul Disord 2008;18:153-155.
71. Nakanishi T, Sakauchi M, Kaneda Y, et al. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006;117:1187-1192.
72. Murakami T, Hayashi YK, Noguchi S, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 2006;60:597-602.
73. Falsaperla R, Romeo G, Mattia C, Pavone P, Quattrocchi S. Subclinical cardiological involvement in Sicilian patients with pure congenital muscular dystrophy. Minerva Pediatr 2005;57:275-279.
74. Ceviz N, Alehan F, Alehan D, et al. Assessment of left ventricular systolic and diastolic functions in children with merosin-positive congenital muscular dystrophy. Int J Cardiol 2003;87:129-133.
75. Gilhuis HJ, ten Donkelaar HJ, Tanke RB, et al. Nonmuscular involvement in merosin-negative congenital muscular dystrophy. Pediatr Neurol 2002;26:30-36.
76. Jones KJ, Morgan G, Johnston H, et al. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet 2001;38:649-657.
77. Finsterer J, Stöllberger C. Primary myopathies and the heart. Scand Cardiovasc J 2008;42:9-24.
78. Perrot A, Spuler S, Geier C, Dietz R, Osterziel KJ. Cardiac manifestations of muscular dystrophies. Z Kardiol 2005;94:312-320.
79. Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging 2007;25:433-440.
80. Peters SA, Köhler C, Schara U, et al. Muscular magnetic resonance imaging for evaluation of myopathies in children. Klin Padiatr 2008;220:37-46.
81. Hillaire D, Leclerc A, Faure S, et al. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum Molec Genet 1994;3:1657-1661.
82. Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin alpha-2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nature Genet 1995;11:216-218.
83. Di Blasi C, Piga D, Brioschi P, et al. LAMA2 gene analysis in congenital muscular dystrophy: new mutations, prenatal diagnosis, and founder effect. Arch Neurol 2005;62:1582-1586.
84. Reed UC, Marie SK, Vainzof M, et al. Congenital muscular dystrophy with cerebral white matter hypodensity. Correlation of clinical features and merosin deficiency.Brain Dev 1996;18:53-58.
85. Vainzof M, Marie SK, Reed UC, et al. Deficiency of merosin (laminin M or alpha 2) in congenital muscular dystrophy associated with cerebral white matter alterations. Neuropediatrics 1995;26:293-297.
86. Leite CC, Lucato LT, Martin MG, et al. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol 2005;35:572-579.
87. Spyrou N, Philpot J, Foale R, Camici PG, Muntoni F. Evidence of left ventricular dysfunction in children with merosin-deficient congenital muscular dystrophy. Am Heart J 1998;136:474-476.
88. Nissinen M, Helbling-Leclerc A, Zhang X, et al. Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am J Hum Genet 1996;58:1177-1184.
89. Herrmann R, Straub V, Meyer K, Kahn T, Wagner M, Voit T. Congenital muscular dystrophy with laminin alpha 2 chain deficiency: identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistry. Eur J Pediatr 1996;155:968-976.
90. Allamand V, Sunada Y, Salih MA, et al. Mild congenital muscular dystrophy in two patients with an internally deleted laminin alpha2-chain. Hum Mol Genet 1997;6:747-752.
91. Sewry CA, Naom I, D'Alessandro M, et al. Variable clinical phenotype in merosin-deficient congenital muscular dystrophy associated with differential immunolabelling of two fragments of the laminin alpha 2 chain. Neuromuscul Disord 1997;7:169-175.
92. Naom I, D'Alessandro M, Sewry C, et al. The role of immunocytochemistry and linkage analysis in the prenatal diagnosis of merosin-deficient congenital muscular dystrophy. Hum Genet 1997;99:535-540.
93. Cohn RD, Herrmann R, Sorokin L, Wewer UM, Voit T. Laminin alpha2 chain-deficient congenital muscular dystrophy: variable epitope expression in severe and mild cases. Neurology 1998;51:94-100.
94. Tezak Z, Prandini P, Boscaro M, et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha-2 (LAMA2) deficiency. Hum Mutat 2003;21:103-111.
95. Shorer Z, Philpot J, Muntoni F, Sewry C, Dubowitz V. Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J. Child Neurol 1995;10:472-475.
96. Quijano-Roy S, Renault F, Romero N, Guicheney P, Fardeau M, Estournet B. EMG and nerve conduction studies in children with congenital muscular dystrophy. Muscle Nerve 2004;29:292-299.
97. Chae JH, Lee JS, Hwang H, et al. Merosin-deficient congenital muscular dystrophy in Korea. Brain Dev 2008 Aug 22 [Epub ahead of print] .
98. Malandrini A, Villanova M, Sabatelli P, et al. Localization of the laminin alpha 2 chain in normal human skeletal muscle and peripheral nerve: an ultrastructural immunolabeling study. Acta Neuropathol 1997;93: 166-172.
99. Villanova M, Malandrini A, Sabatelli P, et al. Localization of laminin alpha 2 chain in normal human central nervous system: an immunofluorescence and ultrastructural study. Acta Neuropathol 1997;94:567-571.
100. Caro PA, Scavina M, Hoffman E, Pegoraro E, Marks HG. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. AJNR Am J Neuroradiol 1999;20:324-326.
101. Leite CC, Reed UC, Otaduy MC, et al. Congenital muscular dystrophy with merosin deficiency: 1H MR spectroscopy and diffusion-weighted MR imaging. Radiology 2005;235:190-196.
102. Sunada Y, Edgar TS, Lotz BP, Rust RS, Campbell KP. Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology 1995;45:2084-2089.
103. Philpot J, Cowan F, Pennock J, et al. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromusc Disord 1999;9:81-85.
104. van der Knaap MS, Smit LM, Barth PG, et al. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol 1997;42:50-59.
105. Vigliano P, Dassi P, Blasi CD, Mora M, Jarre L. LAMA2 stop-codon mutation: Merosin-deficient congenital muscular dystrophy with occipital polymicrogyria, epilepsy and psychomotor regression. Eur J Paediatr Neurol 2008 Apr 11. [Epub ahead of print] .
106. Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 1998;51: 101-110.
107. Sewry CA, D'Alessandro M, Wilson LA, et al. Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics 1997;28:217-222.
108. Siala O, Louhichi N, Triki C, et al. Severe MDC1A congenital muscular dystrophy due to a splicing mutation in the LAMA2 gene resulting in exon skipping and significant decrease of mRNA level. Genet Test 2007;11:199-207.
109. Siala O, Louhichi N, Triki C, Morinière M, Fakhfakh F, Baklouti F. LAMA2 mRNA processing alterations generate a complete deficiency of laminin-alpha2 protein and a severe congenital muscular dystrophy. Neuromuscul Disord 2008;18:137-145.
110. Vainzof M, Richard P, Herrmann R, et al. Prenatal diagnosis in laminin alpha2 chain (merosin)-deficient congenital muscular dystrophy: a collective experienceof five international centers. Neuromuscul Disord 2005;9-10:588-594.
111. Voit T, Fardeau M, Tomé FM. Prenatal detection of merosin expression in human placenta. Neuropediatrics 1994;25:332-333.
112. Talim B, Kale G, Topaloglu H, et al. Clinical and histopathological study of merosin-deficient and merosin-positive congenital muscular dystrophy. Pediatr Dev Pathol 2000;3:168-176.
113. He Y, Jones KJ, Vignier N, et al. Congenital muscular dystrophy with primary partial laminin alpha2 chain deficiency: molecular study. Neurology 2001;57:1319-1322.
114. Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet 2005;42:673-685.
115. Baker NL, Morgelin M, Peat R, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 2005;14:279-293.
116. Baker NL, Mörgelin M, Pace RA, et al. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol 2007;62: 390-405.
117. Demir E, Ferreiro A, Sabatelli P, et al. Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics 2004;35:103-112.
118. Hicks D, Lampe AK, Barresi R, et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology 2008;70:1192-1199.
119. Jimenez-Mallebrera C, Maioli MA, Kim J, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul Disord 2006;16:571-582.
120. Lamande SR, Bateman JF, HutchisonW, et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet 1998;7:981-989.
121. Lamande SR, Shields KA, Kornberg AJ, Shield LK, Bateman JF. Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem 1999;274:21817-21822.
122. Lamande SR, Morgelin M, Selan C, Jobsis GJ, Baas F, Bateman JF. Kinked collagen VI tetramers and reduced microfibril formation as a result of Bethlem myopathy and introduced triple helical glycine mutations. J Biol Chem 2002;277:1949-1956.
123. Lampe AK, Dunn DM, von Niederhausern AC, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet 2005;42:108-120.
124. Lampe AK, Zou Y, Sudano D, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat 2008;29:809-822.
125. Peat RA, Baker NL, Jones KJ, North KN, Lamandé SR. Variable penetrance of COL6A1 null mutations: implications for prenatal diagnosis and genetic counselling in Ullrich congenital muscular dystrophy families. Neuromuscul Disord 2007;17:547-557.
126. Pepe G, Giusti B, Bertini E, et al. A heterozygous splice site mutation in COL6A1 leading to an in-frame deletion of the alpha1(VI) collagen chain in an Italian family affected by Bethlem myopathy. Biochem Biophys Res Commun 1999;258:802-807.
127. Mercuri E, Yuva Y, Brown SC, et al. Collagen VI involvement in Ullrich syndrome: a clinical, genetic, and immunohistochemical study. Neurology 2002;58:1354-1359.
128. Pepe G, Lucarini L, Zhang RZ, et al. COL6A1 genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy. Ann Neurol 2006;59: 190-195.
129. Giusti B, Lucarini L, Pietroni V, et al. Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol 2005;58:400-410.
130. Lucioli S, Giusti B, Mercuri E, et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology 2005;64:1931-1937.
131. Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain 1976;99:91-100.
132. Jobsis GJ, Boers JM, Barth PG, de Visser M. Bethlem myopathy: a slowly progressive congenital muscular dystrophy with contractures. Brain 1999;122:649-655.
133. Reed UC, Ferreira LG, Liu EC, et al. Ullrich congenital muscular dystrophy and Bethlem myopathy: clinical and genetic heterogeneity. Arq Neuropsiquiatr 2005;63:785-790.
134. Schessl J, Goemans NM, Magold AI, et al. Predominant fiber atrophy and fiber type disproportion in early Ullrich disease. Muscle Nerve 2008;38:1184-1191.
135. Kirschner J, Hausser I, Zou Y, et al. Ullrich congenital muscular dystrophy: connective tissue abnormalities in the skin support overlap with Ehlers-Danlos syndromes. Am J Med Genet 2004;132:296-301.
136. Lisi MT, Cohn RD. Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim Biophys Acta 2007;1772:159-172.
137. Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725-2735.
138. Muntoni F. Journey into muscular dystrophies caused by abnormal glycosylation. Acta Myol 2004;23:79-84.
139. Haliloğlu G, Topaloğlu H. Glycosylation defects in muscular dystrophies. Curr Opin Neurol 2004;17:521-527.
140. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Molec Genet 2001;10:2851-2859.
141. Balci B, Uyanik G, Dincer P, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord 2005;15:271-275.
142. Diesen C, Saarinen A, Pihko H, et al. POMGnT1 mutation and phenotypic spectrum in muscle-eye-brain disease. J Med Genet 2004;41:115.
143. Toda T, Kobayashi K, Kondo-Iida E, Sasaki J, Nakamura Y. The Fukuyama congenital muscular dystrophy story. Neuromuscul Disord 2000; 10:153-159.
144. Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis 2006;1:29-36.
145. Mercuri E, Topaloglu H, Brockington M, et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol 2006;63:251-257.
146. Louhichi N, Triki C, Quijano-Roy S, et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 2004;5:27-34.
147. Keramaris-Vrantsis E, Lu PJ, Doran T, et al. Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve 2007;36:455-465.
148. van Reeuwijk J, Maugenre S, van den Elzen C, et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat 2006;27:453-459.
149. Mercuri E, D'Amico A, Tessa A, et al. POMT2 mutation in a patient with 'MEB-like' phenotype. Neuromuscul Disord 2006;16:446-448.
150. Yanagisawa A, Bouchet C, Van den Bergh PY, et al. New POMT2 mutations causing congenital muscular dystrophy: identification of a founder mutation. Neurology 2007;69:1254-1260.
151. Dincer P, Balci B, Yuva Y, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of alpha-dystroglycan. Neuromuscul Disord 2003;13:771-778.
152. D'Amico A, Tessa A, Bruno C, et al. Expanding the clinical spectrum of POMT1 phenotype. Neurology 2006;66:1564-1567.
153. Manya H, Chiba A, Yoshida A, et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA 2004;101:500-505.
154. Messina S, Mora M, Pegoraro E, et al. POMT1 and POMT2 mutations in CMD patients: a multicentric Italian study. Neuromuscul Disord 2008; 18:565-571.
155. Clement EM, Godfrey C, Tan J, et al. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch Neurol 2008; 65:137-141.
156. Lamperti C, Cagliani R, Ciscato P, et al. Congenital muscular dystrophy with muscle inflammation alpha dystroglycan glycosylation defect and no mutation in FKRP gene. J Neurol Sci 2006;243:47-51.
157. Matsumoto H, Hayashi YK, Kim DS, et al. Congenital muscular dystrophy with glycosylation defects of alpha-dystroglycan in Japan. Neuromuscul Disord 2005;15:342-348.
158. Muntoni F, Brockington M, Godfrey C, et al. Muscular dystrophies due to defective glycosylation of dystroglycan. Acta Myol 2007;26:129-135.
159. Cotarelo RP, Valero MC, Prados B, et al. Two new patients bearing mutations in the fukutin gene confirm the relevance of this gene in Walker-Warburg syndrome. Clin Genet 2008;73:139-145.
160. van Reeuwijk J, Brunner HG, van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet 2005;67:281-289.
161. Manzini CM, Gleason D, Chang BS, et al. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mutat 2008;29: E231-E241.
162. Jimenez-Mallebrera C, Torelli S, Feng L, et al. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 2008;Aug 7. [Epub ahead of print] .
163. Zhang W, Vajsar J, Cao P, et al. Enzymatic diagnostic test for Muscle-eye-brain type congenital muscular dystrophy using commercially available reagents. Clin Biochem 2003;36:339-344.
164. Vajsar J, Zhang W, Dobyns WB, et al. Carriers and patients with muscle-eye-brain disease can be rapidly diagnosed by enzymatic analysis of fibroblasts and lymphoblasts. Neuromusc Disord 2006;16:132-136.
165. Manya H, Bouchet C, Yanagisawa A, et al. Protein O-mannosyltransferase activities in lymphoblasts from patients with alpha-dystroglycanopathies. Neuromuscul Disord 2008;18:45-51.
166. Toda T, Chiyonobu T, Xiong H, et al. Fukutin and alpha-dystroglycanopathies. Acta Myol 2005;24:60-63.
167. Toda T, Kobayashi K. Fukuyama-type congenital muscular dystrophy: the first human disease to be caused by an ancient retrotransposal integration. J Mol Med 1999;77:816-823
168. Watanabe M, Kobayashi K, Jin F, et al. Founder SVA retrotransposal insertion in Fukuyama-type congenital muscular dystrophy and its origin in Japanese and Northeast Asian populations. Am J Med Genet 2005;138:344-348.
169. Silan F, Yoshioka M, Kobayashi K, et al. A new mutation of the fukutin gene in a non-Japanese patient. Ann Neurol 2003;53:392-396.
170. Fukuyama Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type--clinical, genetic and pathological considerations. Brain Dev 1981;3:1-30.
171. Osawa M, Sumida S, Suzuki N, et al. Fukuyama type congenital muscular dystrophy. In: Fukuyama Y, Osawa M, Saito K (Eds). Congenital muscular dystrophies. Amsterdam: Elsevier, 1997:31-68.
172. Barkovich AJ. Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol 1998;19:1389-1396.
173. Hino N, Kobayashi M, Shibata N, Yamamoto T, Saito K, Osawa M. Clinicopathological study on eyes from cases of Fukuyama type congenital muscular dystrophy. Brain Dev 2001;23:97-107.
174. Yoshioka M, Kuroki S. Clinical spectrum and genetic studies of Fukuyama congenital muscular dystrophy. Am J Med Genet 1994;53:245-250.
175. Kondo-Iida E, Kobayashi K, Watanabe M, et al. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum Molec Genet 1999; 8:2303-2309.
176. Kondo-Iida E, Saito K, Tanaka H, et al. Molecular genetic evidence of clinical heterogeneity in Fukuyama-type congenital muscular dystrophy. Hum Genet 1997;99:427-432.
177. Saito K, Osawa M, Wang ZP, et al. Haplotype-phenotype correlation in Fukuyama congenital muscular dystrophy. Am J Med Genet 2000; 92:184-190.
178. Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationship in Fukuyama-type congenital muscular dystrophy. Brain Dev 2008;30:59-67.
179. Saito K. Prenatal diagnosis of Fukuyama congenital muscular dystrophy. Prenat Diagn 2006;26:415-417.
180. Raitta C, Santavuori P, Lamminen M, Leisti J. Ophthalmological findings in a new syndrome with muscle, eye and brain involvement. Acta Ophthal 1978;56:465-472.
181. Santavuori P, Somer H, Sainio K, et al. Muscle-eye-brain disease (MEB). Brain Dev 1989;11:147-153.
182. Haltia M, Leivo I, Somer H, et al. Muscle-eye-brain disease: a neuropathological study. Ann Neurol 1997;41:173-180.
183. Leyten QH, Gabreels FJM, Renier WO, Renkawek K, ter Laak HJ, Mullaart RA. Congenital muscular dystrophy with eye and brain malformations in six Dutch patients. Neuropediatrics 1992;23:316-320.
184. Vervoort VS, Holden KR, Ukadike KC, Collins JS, Saul RA, Srivastava AK. POMGnT1 gene alterations in a family with neurological abnormalities. Ann Neurol 2004;56:143-148.
185. Haliloglu G, Gross C, Senbil N,et al. Clinical spectrum of muscle-eye-brain disease: from the typical presentation to severe autistic features. Acta Myol 2004;23:137-139.
186. Hehr U, Uyanik G, Gross C, et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics 2007;8:279-288.
187. Beltran-Valero de Bernabe D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet 2004;41:61.
188. Cormand B, Pihko H, Bayes M, et al. Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology 2001;56:1059-1069.
189. Teber S, Sezer T, Kafali M, et al. Severe muscle-eye-brain disease is associated with a homozygous mutation in the POMGnT1 gene. Eur J Paediatr Neurol 2008;12:133-136.
190. Biancheri R, Bertini E, Falace A, et al. POMGnT1 mutations in congenital muscular dystrophy: genotype-phenotype correlation and expanded clinical spectrum. Arch Neurol 2006;63:1491-1495.
191. Taniguchi K, Kobayashi K, Saito K, et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet 2003;12:527-534.
192. Pagon RA, Chandler JW, Collie WR, et al. Hydrocephalus, agyria, retinal dysplasia, encephalocele (HARD +/- E) syndrome: an autosomal recessive condition. Birth Defects Orig Artic Ser 1978;14:233-241.
193. Dobyns WB, Pagon RA, Armstrong D, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet 1989;32:195-210.
194. Lommel M, Willer T, Strahl S. POMT2, a key enzyme in Walker-Warburg-syndrome: somatic sPOMT2 but not testis specific tPOMT2 is crucial for mannosyltransferase activity in vivo. Glycobiology 2008;18:615-625.
195. Beltran-Valero de Bernabe D, van Bokhoven H, van Beusekom E, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet 2003;40:845-848.
196. van Reeuwijk J, Grewal PK, Salih MA, et al. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet 2007;121: 685-690.
197. Gasser B, Lindner V, Dreyfus M, et al. Prenatal diagnosis of Walker-Warburg syndrome in three sibs. Am J Med Genet 1998;76:107-110.
198. Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 2003;60:988-992.
199. Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 2004;164:727-737.
200. Kirschner J, Bönnemann CG. The congenital and limb-girdle muscular dystrophies: sharpening the focus, blurring the boundaries. Arch Neurol 2004;61:189-199.
201. Driss A, Amouri C, Hamida CB, et al. A new locus for autosomal recessive limb girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromusc Disord 2000;10: 240-246.
202. Godfrey C, Escolar D, Brockington M, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol 2006;60:603-610.
203. Biancheri R, Falace A, Tessa A, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 2007;363:1033-1037.
204. Goebel HH, Lenard H-G, Langenbeck U, Mehl B. A form of congenital muscular dystrophy. Brain Dev 1980;2:387-400.
205. Flanigan KM, Kerr L, Bromberg MB, et al. Congenital muscular dystrophy with rigid spine syndrome: a clinical, pathological, radiological, and genetic study. Ann Neurol 2000;47:152-161.
206. Petit N, Lescure A, Rederstorff M, et al. Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet 2003;12:1045-1053.
207. Tajsharghi H, Darin N, Tulinius M, Oldfors A. Early onset myopathy with a novel mutation in the Selenoprotein N gene (SEPN1). Neuromuscul Disord 2005;15:299-302.
208. Mercuri E, Talim B, Moghadaszadeh B, et al. Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1). Neuromuscul Disord 2002; 12:631-638.
209. Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002;71:739-749.
210. Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol 2004;55:676-686.
211. Fidzianska A, Goebel HH, Osborn M, Lenard HG, Osse G, Langenbeck U. Mallory body-like inclusions in a hereditary congenital neuromuscular disease. Muscle Nerve 1983;6:195-200.
212. Clarke NF, Kidson W, Quijano-Roy S, et al.SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann Neurol 2006;59:546-552.
213. Schara U, Kress W, Bönnemann CG, et al. The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol 2008;12:224-230.
214. Okamoto Y, Takashima H, Higuchi I, et al. Molecular mechanism of rigid spine with muscular dystrophy type 1 caused by novel mutations of selenoprotein N gene. Neurogenetics 2006;7:175-183.
215. Mayer U, Saher G, Fässler R, et al. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet 1997;17:318-323.
216. McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986;319:463-468.
217. Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nature Rev 2006;7:940-952.
218. Worman HJ, Bonne G."Laminopathies": a wide spectrum of human diseases. Exp Cell Res 2007;313:2121-2133.
219. Mercuri E, Brown SC, Nihoyannopoulos P, et al. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve 2005;3:602-609.
220. D'Amico A, Haliloglu G, Richard P, et al. Two patients with 'Dropped head syndrome' due to mutations in LMNA or SEPN1 genes. Neuromuscul Disord 2005;15:521-524.
221. Guglieri M, Magri F, Comi GP. Molecular etiopathogenesis of limb girdle muscular and congenital muscular dystrophies: boundaries and contiguities. Clin Chim Acta 2005;361:54-79.
222. Klein A, Clement E, Mercuri E, Muntoni F. Differential diagnosis of congenital muscular dystrophies. Eur J Paediatr Neurol 2008;12:371-377.
223. Topaloğlu H, Yetük M, Talim B, Akçören Z, Cağlar M. Merosin-positive congenital muscular dystrophy with mental retardation and cataracts: a new entity in two families. Eur J Paediatr Neurol 1997;1:127-131.
224. Reed UC, Tsanaclis AM, Vainzof M, et al. Merosin-positive congenital muscular dystrophy in two siblings with cataract and slight mental retardation. Brain Dev 1999;21:274-278.
225. Reed UC, Marie SK, Vainzof M, et al. Heterogeneity of classic congenital muscular dystrophy with involvement of the central nervous system: report of five atypical cases. J Child Neurol 2000;15:172-178.
226. Mahjneh I, Bushby K, Anderson L, et al. Merosin-positive congenital muscular dystrophy: a large inbred family. Neuropediatrics 1999;30:22-28.
227. Sellick GS, Longman C, Brockington M, et al. Localisation of merosin-positive congenital muscular dystrophy to chromosome 4p16.3. Hum Genet 2005;117:207-212.
228. Tétreault M, Duquette A, Thiffault I, et al. A new form of congenital muscular dystrophy with joint hyperlaxity maps to 3p23-21. Brain 2006; 129:2077-2084.
229. Dohna-Schwake C, Ragette R, Mellies U, Straub V, Teschler H, Voit T. Respiratory function in congenital muscular dystrophy and limb girdle muscular dystrophy 2I. Neurology 2004;62:513-514.
230. Hill NS. Ventilator management for neuromuscular disease. Semin Respir Crit Care Med 2002;23:293-305.
231. Hill NS. Neuromuscular disease in respiratory and critical care medicine. Respir Care 2006;51:1065-1071.
232. Mellies U, Ragette R, Dohna SC, Boehm H, Voit T, Teschler H. Long-term noninvasive ventilation in children and adolescents with neuromuscular disorders. Eur Respir J 2003;22:631-636.
233. Mellies U, Dohna-Schwake C, Voit T. Respiratory function assessment and intervention in neuromuscular disorders. Curr Opin Neurol 2005; 18:543-547.
234. Simonds AK. Respiratory support for the severely handicapped child with neuromuscular disease: ethics and practicality. Semin Respir Crit Care Med 2007;28:342-354.
235. Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A; ENMC. 117th ENMC Workshop: ventilatory support in congenital neuromuscular disorders: congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4-6 April 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:56-69.
236. Weidner NJ. Developing an interdisciplinary palliative care plan for the patient with muscular dystrophy. Pediatr Ann 2005;34:546-552.
237. Alves RS, Resende MB, Skomro RP, Souza FJ, Reed UC. Sleep and neuromuscular disorders in children. Sleep Med Rev 2008 Jun 3. [Epub ahead of print] .
238. Philpot J, Bagnall A, King C, Dubowitz V, Muntoni F. Feeding problems in merosin deficient congenital muscular dystrophy. Arch Dis Child 1999;80:542-547.
239. Ramelli GP, Aloysius A, King C, Davis T, Muntoni F. Gastrostomy placement in paediatric patients with neuromuscular disorders: indications and outcome. Dev Med Child Neurol 2007;49:367-371.
Received 31 October 2008. Accepted 17 December 2008.
Dra. Umbertina Conti Reed Avenida Dr. Enéas de Carvalho Aguiar 255 / 5º andar / sala 5131 - 05403-000 São Paulo SP - Brasil. E-mail: ucontireed@hcnet.usp.br
- 1. Banker BQ, Engel AG. Basic reaction of muscle. Engel AG, Franzini-Armstrong C. Myology: basic and clinical. Edition 3. New York: McGraw-Hill, 2004:691-748.
- 2. Dubowitz V, Sewry CA. Muscle biopsy: a practical approach. Edition 3. Philadelphia: Elsevier, 2006:349-376.
- 3. Mostacciuolo ML, Miorin M, Martinello F, Angelini C, Perini P, Trevisan CP. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet 1996;97:277-279.
- 4. Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord 2000;10:1-9.
- 5. Batten FE. Three cases of myopathy, infantile type. Brain 1903;26:147-148.
- 6. Batten FE. Case of myositis fibrosa with pathological examination. Trans Clin Soc Lond 1904;37:12-22.
- 7. Howard R. A case of congenital defect of the muscular system (Dystrophia muscularis congenita) and its association with congenital talipes equino-varus. Proc R Soc Med 1908;1 Pathol sect:157-166.
- 8. Tomé FM. The Peter Emil Becker Award lecture 1998. The saga of congenital muscular dystrophy. Neuropediatrics 1999;30:55-65.
- Neuromuscul Disord 2003;13:207-215.
- 9. Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14:635-649.
- 10. Voit T, Tome FM. The congenital muscular dystrophies. Engel AG, Franzini-Armstrong C. Myology - basic and clinical. Edition 3. New York: McGraw-Hill-Medical Publishing Division, 2004:1203-1238.
- 11. Ullrich O. Kongenitale atonisch-sklerotische Muskeldystrophie, ein weiterer Typus der heredodegeneration Erkrankungen des nueromuskularen Systems. Z Ges Neurol Psychiat 1930;126:171-201.
- 12. Fukuyama Y, Kawazura M, Haruna H. A peculiar form of congenital progressive muscular dystrophy: report of fifteen cases. Paediat Univ Tokyo 1960;4:5-8.
- 13. Dubowitz V. Rigid spine syndrome: a muscle syndrome in search of a name. Proc R Soc Med 1973;66:219-220.
- 14. Santavuori P, Leisti, J, Kruus S. Muscle, eye and brain disease (MEB): a new syndrome. Neuropädiatrie 1977;8(Suppl):S553-S558.
- 15. Walker AE. Lissencephaly. Arch Neurol Psychiatr 1942:48:13-29.
- 16. Warburg M. Heterogeneity of congenital retinal nonattachment, falciform folds and retinal dysplasia: a guide to genetic counselling. Hum Hered 1976:26:137-148.
- 17. Dobyns WB, Pagon RA, Armstrong D, et al. New diagnostic criteria for Walker-Warburg syndrome. Am J Hum Genet 1986;39:A59.
- 18. Dubowitz V. 22nd ENMC sponsored workshop on congenital muscular dystrophy held in Baarn, The Netherlands, 14-16 May 1993. Neuromuscul Disord 1994;4:75-81.
- 19. Kanagawa M, Saito F, Kunz S, et al. Congenital muscular dystrophy of a non-Fukuyama type with white matter hyperlucency on CT scan. Brain Dev 1992;14:420-422.
- 20. Kao KP, Lin KP. Congenital muscular dystrophy of a non-Fukuyama type with white matter hyperlucency on CT scan.Brain Dev 1992;14:420-422.
- 21. Oliveira AS, Gabbai AA, Kiyomoto BH, Schmidt B. Congenital muscular dystrophy: report of 8 cases with evidence of central nervous system involvement. Rev Paul Med 1990;108:139-141.
- 22. Oliveira AS, Gabbai AA, Kiyomoto BH, Ferreira Neto A, Schmidt B, Lima JG. Congenital muscular dystrophy: clinical study of 17 patients. Arq Neuropsiquiatr 1991;49:265-271.
- 23. Tanaka J, Mimaki T, Okada S, Fujimura H. Changes in cerebral white matter in a case of congenital muscular dystrophy (non-Fukuyama type). Neuropediatrics 1990;21:183-186.
- 24. Topaloglu H, Yalaz K, Kale G, Ergin M. Congenital muscular dystrophy with cerebral involvement: report of a case of "occidental type cerebromuscular dystrophy"? Neuropediatrics 1990;21:53-54.
- 25. Trevisan CP, Carollo C, Segalla P, Angelini C, Drigo P, Giordano R. Congenital muscular dystrophy: brain alterations in an unselected series of Western patients. J Neurol Neurosurg Psychiatry 1991;54:330-334.
- 26. Toda T, Kanazawa I, Nakamura Y. Localization of a gene responsible for Fukuyama type congenital muscular dystrophy to chromosome 9q31-33 by linkage analysis. Human Genome Mapping Workshop 1993:A20.
- 27. Tome FM, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci 1994;317:351-357.
- 28. Burgeson RE, Chiquet M, Deutzmann R, et al. A new nomenclature for the laminins. Matrix Biol 1994;14:209-211.
- 29. Kobayashi K, Nakahori Y. Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998;394:388-392.
- 30. Moghadaszadeh B, Desguerre I, Topaloglu H, et al. Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigidity of the spine, on chromosome 1p35-36. Am J Hum Genet 1998;62:1439-1445.
- 31. Muntoni F, Taylor J, Sewry CA, Naom I, Dubowitz V. An early onset muscular dystrophy with diaphragmatic involvement, early respiratory failure and secondary alpha2 laminin deficiency unlinked to the LAMA2 locus on 6q22. Eur J Paediatr Neurol 1998;1:19-26.
- 32. Cormand B, Avela K, Pihko H, et al. Assignment of the muscle-eye-brain disease gene to 1p32-p34 by linkage analysis and homozygosity mapping. Am J Hum Genet 1999;64:126-135.
- 33. Brockington M, Sewry CA, Herrmann R, et al. Assignment of a form of congenital muscular dystrophy with secondary merosin deficiency to chromosome 1q42. Am J Hum Genet 2000;66:428-435.
- 34. Moghadaszadeh B, Petit N, Jaillard C, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nature Genet 2001;29:17-18.
- 35. Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA 2001;98:7516-7521.
- 36. Demir E, Sabatelli P, Allamand V, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet 2002;70:1446-1458.
- 37. Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet 2003;73:355-369.
- 38. Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha-2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001;69:1198-1209.
- 39. Hayashi YK, Ogawa M, Tagawa K, et al. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001;57:115-121.
- 40. Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001;1:717-724.
- 41. Kano H, Kobayashi K, Herrmann R, et al. Deficiency of alpha-dystroglycan in muscle-eye-brain disease. Biochem Biophys Res Commun 2002;291:1283-1286.
- 42. Michele DE, Barresi R, Kanagawa M, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002;418:417-422.
- 43. Moore SA, Saito F, Chen J, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002;418: 422-425.
- 44. Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet 2002;360:1419-1421.
- 45. Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71:1033-1043.
- 46. Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Molec Genet 2003;12:2853-2861.
- 47. van Reeuwijk J, Janssen M, van den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 2005;42:907-912.
- 48. Dubowitz V. 68th ENMC International Workshop (5th International Workshop): on Congenital Muscular Dystrophy, 9-11 April 1999, Naarden, The Netherlands. Neuromuscul Disord 1999;9:446-454.
- 49. Muntoni F, Guicheney P. 85th ENMC International Workshop on Congenital Muscular Dystrophy. 6th International CMD Workshop. 1st Workshop of the Myo-Cluster Project 'GENRE'. 27-28th October 2000, Naarden, The Netherlands. Neuromuscul Disord 2002;12:69-78.
- 50. Muntoni F, Bertini E, Bönnemann C, et al. 98th ENMC International Workshop on Congenital Muscular Dystrophy (CMD), 7th Workshop of the International Consortium on CMD, 2nd Workshop of the MYO CLUSTER project GENRE. 26-28th October, 2001, Naarden, The Netherlands. Neuromuscul Disord 2002;12:889-896.
- 51. Muntoni F, Valero de Bernabe B, Bittner R, et al. 114th ENMC International Workshop on Congenital Muscular Dystrophy (CMD) 17-19 January 2003, Naarden, The Netherlands: (8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE). Neuromuscul Disord 2003;13:579-588.
- 52. Muntoni F, Voit T. 133rd ENMC International Workshop on Congenital Muscular Dystrophy (IXth International CMD Workshop) 21-23 January 2005, Naarden, The Netherlands. Neuromuscul Disord 2005;15:794-801.
- 53. Fukuyama Y. Fukuyama congenital muscular dystrophy-history and perspectives. Brain Nerve 2008;60:43-51.
- 54. Gene table of monogenic neuromuscular disorders (nuclear genome only). Neuromusc Disord 2008;18:101-129.
- 55. Quijano-Roy S, Mbieleu B, Bönnemann CG, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008;64:177-186.
- 56. Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F. Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 2005;62:809-823.
- 57. Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006;9:427-443.
- 58. Ferreira LG, Marie SK, Liu EC, et al. Dystrophin-glycoproteins associated in congenital muscular dystrophy: immunohistochemical analysis of 59 Brazilian cases. Arq Neuropsiquiatr 2005;63:791-800.
- 59. Pepe G, Bertini E, Bonaldo P, et al. Bethlem myopathy (BETHLEM) and Ullrich Scleroatonic Muscular Dystrophy: 100th ENMC International Workshop, 23-24 November 2001, Naarden, The Netherlands. Neuromuscul Disord 2002;12:984-993.
- 60. Okada M, Kawahara G, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007;69:1035-1042.
- 61. Peat RA, Smith JM, Compton AG, et al. The diagnosis and etiology of congenital muscular dystrophy. Neurology 2008;71:312-321.
- 62. Mercuri E, Longman C. Congenital muscular dystrophy. Pediatr Ann 2005;34:560-562.
- 63. Hayashi YK, ChouFL, Engvall E, et al. Mutations in the integrin alpha-7 gene cause congenital myopathy. Nature Genet 1998;19:94-97.
- 64. Merlini L, Martoni E, Grumati P, et al. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 2008;71:1245-1253.
- 65. Jones KJ, North KN. External ophthalmoplegia in neuromuscular disorders: case report and review of the literature. Neuromuscul Disord 1997;7:143-151.
- 66. Salih MA, Al Rayess M, Cutshall S, et al. A novel form of familial congenital muscular dystrophy in two adolescents. Neuropediatrics 1998; 29:289-293.
- 67. Voit T, Parano E, Straub V, et al. Congenital muscular dystrophy with adducted thumbs, ptosis, external ophthalmoplegia, mental retardation and cerebellar hypoplasia: a novel form of CMD. Neuromuscul Disord 2002;12:623-630.
- 68. Jones KJ, Compton AG, Yang N, et al. Deficiency of the syntrophins and alpha-dystrobrevin in patients with inherited myopathy. Neuromuscul Disord 2003;13:456-467.
- 69. Vondracek P, Hermanova M, Vodickova K, et al. An unusual case of congenital muscular dystrophy with normal serum CK level, external ophtalmoplegia, and white matter changes on brain MRI. Eur J Paediatr Neurol 2007;11:381-384.
- 70. D'Amico A, Petrini S, Parisi F, et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul Disord 2008;18:153-155.
- 71. Nakanishi T, Sakauchi M, Kaneda Y, et al. Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006;117:1187-1192.
- 72. Murakami T, Hayashi YK, Noguchi S, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 2006;60:597-602.
- 73. Falsaperla R, Romeo G, Mattia C, Pavone P, Quattrocchi S. Subclinical cardiological involvement in Sicilian patients with pure congenital muscular dystrophy. Minerva Pediatr 2005;57:275-279.
- 74. Ceviz N, Alehan F, Alehan D, et al. Assessment of left ventricular systolic and diastolic functions in children with merosin-positive congenital muscular dystrophy. Int J Cardiol 2003;87:129-133.
- 75. Gilhuis HJ, ten Donkelaar HJ, Tanke RB, et al. Nonmuscular involvement in merosin-negative congenital muscular dystrophy. Pediatr Neurol 2002;26:30-36.
- 76. Jones KJ, Morgan G, Johnston H, et al. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J Med Genet 2001;38:649-657.
- 77. Finsterer J, Stöllberger C. Primary myopathies and the heart. Scand Cardiovasc J 2008;42:9-24.
- 78. Perrot A, Spuler S, Geier C, Dietz R, Osterziel KJ. Cardiac manifestations of muscular dystrophies. Z Kardiol 2005;94:312-320.
- 79. Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging 2007;25:433-440.
- 80. Peters SA, Köhler C, Schara U, et al. Muscular magnetic resonance imaging for evaluation of myopathies in children. Klin Padiatr 2008;220:37-46.
- 81. Hillaire D, Leclerc A, Faure S, et al. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum Molec Genet 1994;3:1657-1661.
- 82. Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin alpha-2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nature Genet 1995;11:216-218.
- 83. Di Blasi C, Piga D, Brioschi P, et al. LAMA2 gene analysis in congenital muscular dystrophy: new mutations, prenatal diagnosis, and founder effect. Arch Neurol 2005;62:1582-1586.
- 84. Reed UC, Marie SK, Vainzof M, et al. Congenital muscular dystrophy with cerebral white matter hypodensity. Correlation of clinical features and merosin deficiency.Brain Dev 1996;18:53-58.
- 85. Vainzof M, Marie SK, Reed UC, et al. Deficiency of merosin (laminin M or alpha 2) in congenital muscular dystrophy associated with cerebral white matter alterations. Neuropediatrics 1995;26:293-297.
- 86. Leite CC, Lucato LT, Martin MG, et al. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol 2005;35:572-579.
- 87. Spyrou N, Philpot J, Foale R, Camici PG, Muntoni F. Evidence of left ventricular dysfunction in children with merosin-deficient congenital muscular dystrophy. Am Heart J 1998;136:474-476.
- 88. Nissinen M, Helbling-Leclerc A, Zhang X, et al. Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am J Hum Genet 1996;58:1177-1184.
- 89. Herrmann R, Straub V, Meyer K, Kahn T, Wagner M, Voit T. Congenital muscular dystrophy with laminin alpha 2 chain deficiency: identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistry. Eur J Pediatr 1996;155:968-976.
- 90. Allamand V, Sunada Y, Salih MA, et al. Mild congenital muscular dystrophy in two patients with an internally deleted laminin alpha2-chain. Hum Mol Genet 1997;6:747-752.
- 91. Sewry CA, Naom I, D'Alessandro M, et al. Variable clinical phenotype in merosin-deficient congenital muscular dystrophy associated with differential immunolabelling of two fragments of the laminin alpha 2 chain. Neuromuscul Disord 1997;7:169-175.
- 92. Naom I, D'Alessandro M, Sewry C, et al. The role of immunocytochemistry and linkage analysis in the prenatal diagnosis of merosin-deficient congenital muscular dystrophy. Hum Genet 1997;99:535-540.
- 93. Cohn RD, Herrmann R, Sorokin L, Wewer UM, Voit T. Laminin alpha2 chain-deficient congenital muscular dystrophy: variable epitope expression in severe and mild cases. Neurology 1998;51:94-100.
- 94. Tezak Z, Prandini P, Boscaro M, et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha-2 (LAMA2) deficiency. Hum Mutat 2003;21:103-111.
- 95. Shorer Z, Philpot J, Muntoni F, Sewry C, Dubowitz V. Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J. Child Neurol 1995;10:472-475.
- 96. Quijano-Roy S, Renault F, Romero N, Guicheney P, Fardeau M, Estournet B. EMG and nerve conduction studies in children with congenital muscular dystrophy. Muscle Nerve 2004;29:292-299.
- 97. Chae JH, Lee JS, Hwang H, et al. Merosin-deficient congenital muscular dystrophy in Korea. Brain Dev 2008 Aug 22 [Epub ahead of print]
- 98. Malandrini A, Villanova M, Sabatelli P, et al. Localization of the laminin alpha 2 chain in normal human skeletal muscle and peripheral nerve: an ultrastructural immunolabeling study. Acta Neuropathol 1997;93: 166-172.
- 99. Villanova M, Malandrini A, Sabatelli P, et al. Localization of laminin alpha 2 chain in normal human central nervous system: an immunofluorescence and ultrastructural study. Acta Neuropathol 1997;94:567-571.
- 100. Caro PA, Scavina M, Hoffman E, Pegoraro E, Marks HG. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. AJNR Am J Neuroradiol 1999;20:324-326.
- 101. Leite CC, Reed UC, Otaduy MC, et al. Congenital muscular dystrophy with merosin deficiency: 1H MR spectroscopy and diffusion-weighted MR imaging. Radiology 2005;235:190-196.
- 102. Sunada Y, Edgar TS, Lotz BP, Rust RS, Campbell KP. Merosin-negative congenital muscular dystrophy associated with extensive brain abnormalities. Neurology 1995;45:2084-2089.
- 103. Philpot J, Cowan F, Pennock J, et al. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromusc Disord 1999;9:81-85.
- 104. van der Knaap MS, Smit LM, Barth PG, et al. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann Neurol 1997;42:50-59.
- 105. Vigliano P, Dassi P, Blasi CD, Mora M, Jarre L. LAMA2 stop-codon mutation: Merosin-deficient congenital muscular dystrophy with occipital polymicrogyria, epilepsy and psychomotor regression. Eur J Paediatr Neurol 2008 Apr 11. [Epub ahead of print]
- 106. Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 1998;51: 101-110.
- 107. Sewry CA, D'Alessandro M, Wilson LA, et al. Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics 1997;28:217-222.
- 108. Siala O, Louhichi N, Triki C, et al. Severe MDC1A congenital muscular dystrophy due to a splicing mutation in the LAMA2 gene resulting in exon skipping and significant decrease of mRNA level. Genet Test 2007;11:199-207.
- 109. Siala O, Louhichi N, Triki C, Morinière M, Fakhfakh F, Baklouti F. LAMA2 mRNA processing alterations generate a complete deficiency of laminin-alpha2 protein and a severe congenital muscular dystrophy. Neuromuscul Disord 2008;18:137-145.
- 110. Vainzof M, Richard P, Herrmann R, et al. Prenatal diagnosis in laminin alpha2 chain (merosin)-deficient congenital muscular dystrophy: a collective experienceof five international centers. Neuromuscul Disord 2005;9-10:588-594.
- 111. Voit T, Fardeau M, Tomé FM. Prenatal detection of merosin expression in human placenta. Neuropediatrics 1994;25:332-333.
- 112. Talim B, Kale G, Topaloglu H, et al. Clinical and histopathological study of merosin-deficient and merosin-positive congenital muscular dystrophy. Pediatr Dev Pathol 2000;3:168-176.
- 113. He Y, Jones KJ, Vignier N, et al. Congenital muscular dystrophy with primary partial laminin alpha2 chain deficiency: molecular study. Neurology 2001;57:1319-1322.
- 114. Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet 2005;42:673-685.
- 115. Baker NL, Morgelin M, Peat R, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 2005;14:279-293.
- 116. Baker NL, Mörgelin M, Pace RA, et al. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol 2007;62: 390-405.
- 117. Demir E, Ferreiro A, Sabatelli P, et al. Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics 2004;35:103-112.
- 118. Hicks D, Lampe AK, Barresi R, et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology 2008;70:1192-1199.
- 119. Jimenez-Mallebrera C, Maioli MA, Kim J, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul Disord 2006;16:571-582.
- 120. Lamande SR, Bateman JF, HutchisonW, et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet 1998;7:981-989.
- 121. Lamande SR, Shields KA, Kornberg AJ, Shield LK, Bateman JF. Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem 1999;274:21817-21822.
- 122. Lamande SR, Morgelin M, Selan C, Jobsis GJ, Baas F, Bateman JF. Kinked collagen VI tetramers and reduced microfibril formation as a result of Bethlem myopathy and introduced triple helical glycine mutations. J Biol Chem 2002;277:1949-1956.
- 123. Lampe AK, Dunn DM, von Niederhausern AC, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet 2005;42:108-120.
- 124. Lampe AK, Zou Y, Sudano D, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat 2008;29:809-822.
- 125. Peat RA, Baker NL, Jones KJ, North KN, Lamandé SR. Variable penetrance of COL6A1 null mutations: implications for prenatal diagnosis and genetic counselling in Ullrich congenital muscular dystrophy families. Neuromuscul Disord 2007;17:547-557.
- 126. Pepe G, Giusti B, Bertini E, et al. A heterozygous splice site mutation in COL6A1 leading to an in-frame deletion of the alpha1(VI) collagen chain in an Italian family affected by Bethlem myopathy. Biochem Biophys Res Commun 1999;258:802-807.
- 127. Mercuri E, Yuva Y, Brown SC, et al. Collagen VI involvement in Ullrich syndrome: a clinical, genetic, and immunohistochemical study. Neurology 2002;58:1354-1359.
- 128. Pepe G, Lucarini L, Zhang RZ, et al. COL6A1 genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy. Ann Neurol 2006;59: 190-195.
- 129. Giusti B, Lucarini L, Pietroni V, et al. Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol 2005;58:400-410.
- 130. Lucioli S, Giusti B, Mercuri E, et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology 2005;64:1931-1937.
- 131. Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain 1976;99:91-100.
- 132. Jobsis GJ, Boers JM, Barth PG, de Visser M. Bethlem myopathy: a slowly progressive congenital muscular dystrophy with contractures. Brain 1999;122:649-655.
- 133. Reed UC, Ferreira LG, Liu EC, et al. Ullrich congenital muscular dystrophy and Bethlem myopathy: clinical and genetic heterogeneity. Arq Neuropsiquiatr 2005;63:785-790.
- 134. Schessl J, Goemans NM, Magold AI, et al. Predominant fiber atrophy and fiber type disproportion in early Ullrich disease. Muscle Nerve 2008;38:1184-1191.
- 135. Kirschner J, Hausser I, Zou Y, et al. Ullrich congenital muscular dystrophy: connective tissue abnormalities in the skin support overlap with Ehlers-Danlos syndromes. Am J Med Genet 2004;132:296-301.
- 136. Lisi MT, Cohn RD. Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim Biophys Acta 2007;1772:159-172.
- 137. Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725-2735.
- 138. Muntoni F. Journey into muscular dystrophies caused by abnormal glycosylation. Acta Myol 2004;23:79-84.
- 140. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Molec Genet 2001;10:2851-2859.
- 141. Balci B, Uyanik G, Dincer P, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord 2005;15:271-275.
- 142. Diesen C, Saarinen A, Pihko H, et al. POMGnT1 mutation and phenotypic spectrum in muscle-eye-brain disease. J Med Genet 2004;41:115.
- 143. Toda T, Kobayashi K, Kondo-Iida E, Sasaki J, Nakamura Y. The Fukuyama congenital muscular dystrophy story. Neuromuscul Disord 2000; 10:153-159.
- 144. Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis 2006;1:29-36.
- 145. Mercuri E, Topaloglu H, Brockington M, et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol 2006;63:251-257.
- 146. Louhichi N, Triki C, Quijano-Roy S, et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 2004;5:27-34.
- 147. Keramaris-Vrantsis E, Lu PJ, Doran T, et al. Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve 2007;36:455-465.
- 148. van Reeuwijk J, Maugenre S, van den Elzen C, et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat 2006;27:453-459.
- 149. Mercuri E, D'Amico A, Tessa A, et al. POMT2 mutation in a patient with 'MEB-like' phenotype. Neuromuscul Disord 2006;16:446-448.
- 150. Yanagisawa A, Bouchet C, Van den Bergh PY, et al. New POMT2 mutations causing congenital muscular dystrophy: identification of a founder mutation. Neurology 2007;69:1254-1260.
- 151. Dincer P, Balci B, Yuva Y, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of alpha-dystroglycan. Neuromuscul Disord 2003;13:771-778.
- 152. D'Amico A, Tessa A, Bruno C, et al. Expanding the clinical spectrum of POMT1 phenotype. Neurology 2006;66:1564-1567.
- 153. Manya H, Chiba A, Yoshida A, et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA 2004;101:500-505.
- 154. Messina S, Mora M, Pegoraro E, et al. POMT1 and POMT2 mutations in CMD patients: a multicentric Italian study. Neuromuscul Disord 2008; 18:565-571.
- 155. Clement EM, Godfrey C, Tan J, et al. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch Neurol 2008; 65:137-141.
- 156. Lamperti C, Cagliani R, Ciscato P, et al. Congenital muscular dystrophy with muscle inflammation alpha dystroglycan glycosylation defect and no mutation in FKRP gene. J Neurol Sci 2006;243:47-51.
- 157. Matsumoto H, Hayashi YK, Kim DS, et al. Congenital muscular dystrophy with glycosylation defects of alpha-dystroglycan in Japan. Neuromuscul Disord 2005;15:342-348.
- 158. Muntoni F, Brockington M, Godfrey C, et al. Muscular dystrophies due to defective glycosylation of dystroglycan. Acta Myol 2007;26:129-135.
- 159. Cotarelo RP, Valero MC, Prados B, et al. Two new patients bearing mutations in the fukutin gene confirm the relevance of this gene in Walker-Warburg syndrome. Clin Genet 2008;73:139-145.
- 160. van Reeuwijk J, Brunner HG, van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet 2005;67:281-289.
- 161. Manzini CM, Gleason D, Chang BS, et al. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mutat 2008;29: E231-E241.
- 162. Jimenez-Mallebrera C, Torelli S, Feng L, et al. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 2008;Aug 7. [Epub ahead of print]
- 163. Zhang W, Vajsar J, Cao P, et al. Enzymatic diagnostic test for Muscle-eye-brain type congenital muscular dystrophy using commercially available reagents. Clin Biochem 2003;36:339-344.
- 164. Vajsar J, Zhang W, Dobyns WB, et al. Carriers and patients with muscle-eye-brain disease can be rapidly diagnosed by enzymatic analysis of fibroblasts and lymphoblasts. Neuromusc Disord 2006;16:132-136.
- 165. Manya H, Bouchet C, Yanagisawa A, et al. Protein O-mannosyltransferase activities in lymphoblasts from patients with alpha-dystroglycanopathies. Neuromuscul Disord 2008;18:45-51.
- 166. Toda T, Chiyonobu T, Xiong H, et al. Fukutin and alpha-dystroglycanopathies. Acta Myol 2005;24:60-63.
- 167. Toda T, Kobayashi K. Fukuyama-type congenital muscular dystrophy: the first human disease to be caused by an ancient retrotransposal integration. J Mol Med 1999;77:816-823
- 168. Watanabe M, Kobayashi K, Jin F, et al. Founder SVA retrotransposal insertion in Fukuyama-type congenital muscular dystrophy and its origin in Japanese and Northeast Asian populations. Am J Med Genet 2005;138:344-348.
- 169. Silan F, Yoshioka M, Kobayashi K, et al. A new mutation of the fukutin gene in a non-Japanese patient. Ann Neurol 2003;53:392-396.
- 170. Fukuyama Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type--clinical, genetic and pathological considerations. Brain Dev 1981;3:1-30.
- 171. Osawa M, Sumida S, Suzuki N, et al. Fukuyama type congenital muscular dystrophy. In: Fukuyama Y, Osawa M, Saito K (Eds). Congenital muscular dystrophies. Amsterdam: Elsevier, 1997:31-68.
- 172. Barkovich AJ. Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol 1998;19:1389-1396.
- 173. Hino N, Kobayashi M, Shibata N, Yamamoto T, Saito K, Osawa M. Clinicopathological study on eyes from cases of Fukuyama type congenital muscular dystrophy. Brain Dev 2001;23:97-107.
- 174. Yoshioka M, Kuroki S. Clinical spectrum and genetic studies of Fukuyama congenital muscular dystrophy. Am J Med Genet 1994;53:245-250.
- 175. Kondo-Iida E, Kobayashi K, Watanabe M, et al. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD). Hum Molec Genet 1999; 8:2303-2309.
- 176. Kondo-Iida E, Saito K, Tanaka H, et al. Molecular genetic evidence of clinical heterogeneity in Fukuyama-type congenital muscular dystrophy. Hum Genet 1997;99:427-432.
- 177. Saito K, Osawa M, Wang ZP, et al. Haplotype-phenotype correlation in Fukuyama congenital muscular dystrophy. Am J Med Genet 2000; 92:184-190.
- 178. Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationship in Fukuyama-type congenital muscular dystrophy. Brain Dev 2008;30:59-67.
- 179. Saito K. Prenatal diagnosis of Fukuyama congenital muscular dystrophy. Prenat Diagn 2006;26:415-417.
- 180. Raitta C, Santavuori P, Lamminen M, Leisti J. Ophthalmological findings in a new syndrome with muscle, eye and brain involvement. Acta Ophthal 1978;56:465-472.
- 181. Santavuori P, Somer H, Sainio K, et al. Muscle-eye-brain disease (MEB). Brain Dev 1989;11:147-153.
- 182. Haltia M, Leivo I, Somer H, et al. Muscle-eye-brain disease: a neuropathological study. Ann Neurol 1997;41:173-180.
- 183. Leyten QH, Gabreels FJM, Renier WO, Renkawek K, ter Laak HJ, Mullaart RA. Congenital muscular dystrophy with eye and brain malformations in six Dutch patients. Neuropediatrics 1992;23:316-320.
- 184. Vervoort VS, Holden KR, Ukadike KC, Collins JS, Saul RA, Srivastava AK. POMGnT1 gene alterations in a family with neurological abnormalities. Ann Neurol 2004;56:143-148.
- 185. Haliloglu G, Gross C, Senbil N,et al. Clinical spectrum of muscle-eye-brain disease: from the typical presentation to severe autistic features. Acta Myol 2004;23:137-139.
- 186. Hehr U, Uyanik G, Gross C, et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics 2007;8:279-288.
- 187. Beltran-Valero de Bernabe D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet 2004;41:61.
- 188. Cormand B, Pihko H, Bayes M, et al. Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology 2001;56:1059-1069.
- 189. Teber S, Sezer T, Kafali M, et al. Severe muscle-eye-brain disease is associated with a homozygous mutation in the POMGnT1 gene. Eur J Paediatr Neurol 2008;12:133-136.
- 190. Biancheri R, Bertini E, Falace A, et al. POMGnT1 mutations in congenital muscular dystrophy: genotype-phenotype correlation and expanded clinical spectrum. Arch Neurol 2006;63:1491-1495.
- 191. Taniguchi K, Kobayashi K, Saito K, et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet 2003;12:527-534.
- 192. Pagon RA, Chandler JW, Collie WR, et al. Hydrocephalus, agyria, retinal dysplasia, encephalocele (HARD +/- E) syndrome: an autosomal recessive condition. Birth Defects Orig Artic Ser 1978;14:233-241.
- 193. Dobyns WB, Pagon RA, Armstrong D, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet 1989;32:195-210.
- 194. Lommel M, Willer T, Strahl S. POMT2, a key enzyme in Walker-Warburg-syndrome: somatic sPOMT2 but not testis specific tPOMT2 is crucial for mannosyltransferase activity in vivo. Glycobiology 2008;18:615-625.
- 195. Beltran-Valero de Bernabe D, van Bokhoven H, van Beusekom E, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet 2003;40:845-848.
- 196. van Reeuwijk J, Grewal PK, Salih MA, et al. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet 2007;121: 685-690.
- 197. Gasser B, Lindner V, Dreyfus M, et al. Prenatal diagnosis of Walker-Warburg syndrome in three sibs. Am J Med Genet 1998;76:107-110.
- 198. Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 2003;60:988-992.
- 199. Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 2004;164:727-737.
- 200. Kirschner J, Bönnemann CG. The congenital and limb-girdle muscular dystrophies: sharpening the focus, blurring the boundaries. Arch Neurol 2004;61:189-199.
- 201. Driss A, Amouri C, Hamida CB, et al. A new locus for autosomal recessive limb girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromusc Disord 2000;10: 240-246.
- 202. Godfrey C, Escolar D, Brockington M, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol 2006;60:603-610.
- 203. Biancheri R, Falace A, Tessa A, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 2007;363:1033-1037.
- 204. Goebel HH, Lenard H-G, Langenbeck U, Mehl B. A form of congenital muscular dystrophy. Brain Dev 1980;2:387-400.
- 205. Flanigan KM, Kerr L, Bromberg MB, et al. Congenital muscular dystrophy with rigid spine syndrome: a clinical, pathological, radiological, and genetic study. Ann Neurol 2000;47:152-161.
- 206. Petit N, Lescure A, Rederstorff M, et al. Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet 2003;12:1045-1053.
- 207. Tajsharghi H, Darin N, Tulinius M, Oldfors A. Early onset myopathy with a novel mutation in the Selenoprotein N gene (SEPN1). Neuromuscul Disord 2005;15:299-302.
- 208. Mercuri E, Talim B, Moghadaszadeh B, et al. Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1). Neuromuscul Disord 2002; 12:631-638.
- 209. Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002;71:739-749.
- 210. Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol 2004;55:676-686.
- 211. Fidzianska A, Goebel HH, Osborn M, Lenard HG, Osse G, Langenbeck U. Mallory body-like inclusions in a hereditary congenital neuromuscular disease. Muscle Nerve 1983;6:195-200.
- 212. Clarke NF, Kidson W, Quijano-Roy S, et al.SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann Neurol 2006;59:546-552.
- 213. Schara U, Kress W, Bönnemann CG, et al. The phenotype and long-term follow-up in 11 patients with juvenile selenoprotein N1-related myopathy. Eur J Paediatr Neurol 2008;12:224-230.
- 214. Okamoto Y, Takashima H, Higuchi I, et al. Molecular mechanism of rigid spine with muscular dystrophy type 1 caused by novel mutations of selenoprotein N gene. Neurogenetics 2006;7:175-183.
- 215. Mayer U, Saher G, Fässler R, et al. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet 1997;17:318-323.
- 216. McKeon FD, Kirschner MW, Caput D. Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 1986;319:463-468.
- 217. Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nature Rev 2006;7:940-952.
- 218. Worman HJ, Bonne G."Laminopathies": a wide spectrum of human diseases. Exp Cell Res 2007;313:2121-2133.
- 219. Mercuri E, Brown SC, Nihoyannopoulos P, et al. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve 2005;3:602-609.
- 220. D'Amico A, Haliloglu G, Richard P, et al. Two patients with 'Dropped head syndrome' due to mutations in LMNA or SEPN1 genes. Neuromuscul Disord 2005;15:521-524.
- 221. Guglieri M, Magri F, Comi GP. Molecular etiopathogenesis of limb girdle muscular and congenital muscular dystrophies: boundaries and contiguities. Clin Chim Acta 2005;361:54-79.
- 222. Klein A, Clement E, Mercuri E, Muntoni F. Differential diagnosis of congenital muscular dystrophies. Eur J Paediatr Neurol 2008;12:371-377.
- 224. Reed UC, Tsanaclis AM, Vainzof M, et al. Merosin-positive congenital muscular dystrophy in two siblings with cataract and slight mental retardation. Brain Dev 1999;21:274-278.
- 225. Reed UC, Marie SK, Vainzof M, et al. Heterogeneity of classic congenital muscular dystrophy with involvement of the central nervous system: report of five atypical cases. J Child Neurol 2000;15:172-178.
- 226. Mahjneh I, Bushby K, Anderson L, et al. Merosin-positive congenital muscular dystrophy: a large inbred family. Neuropediatrics 1999;30:22-28.
- 227. Sellick GS, Longman C, Brockington M, et al. Localisation of merosin-positive congenital muscular dystrophy to chromosome 4p16.3. Hum Genet 2005;117:207-212.
- 228. Tétreault M, Duquette A, Thiffault I, et al. A new form of congenital muscular dystrophy with joint hyperlaxity maps to 3p23-21. Brain 2006; 129:2077-2084.
- 229. Dohna-Schwake C, Ragette R, Mellies U, Straub V, Teschler H, Voit T. Respiratory function in congenital muscular dystrophy and limb girdle muscular dystrophy 2I. Neurology 2004;62:513-514.
- 230. Hill NS. Ventilator management for neuromuscular disease. Semin Respir Crit Care Med 2002;23:293-305.
- 231. Hill NS. Neuromuscular disease in respiratory and critical care medicine. Respir Care 2006;51:1065-1071.
- 232. Mellies U, Ragette R, Dohna SC, Boehm H, Voit T, Teschler H. Long-term noninvasive ventilation in children and adolescents with neuromuscular disorders. Eur Respir J 2003;22:631-636.
- 233. Mellies U, Dohna-Schwake C, Voit T. Respiratory function assessment and intervention in neuromuscular disorders. Curr Opin Neurol 2005; 18:543-547.
- 234. Simonds AK. Respiratory support for the severely handicapped child with neuromuscular disease: ethics and practicality. Semin Respir Crit Care Med 2007;28:342-354.
- 235. Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A; ENMC. 117th ENMC Workshop: ventilatory support in congenital neuromuscular disorders: congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4-6 April 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:56-69.
- 236. Weidner NJ. Developing an interdisciplinary palliative care plan for the patient with muscular dystrophy. Pediatr Ann 2005;34:546-552.
- 237. Alves RS, Resende MB, Skomro RP, Souza FJ, Reed UC. Sleep and neuromuscular disorders in children. Sleep Med Rev 2008 Jun 3. [Epub ahead of print]
- 238. Philpot J, Bagnall A, King C, Dubowitz V, Muntoni F. Feeding problems in merosin deficient congenital muscular dystrophy. Arch Dis Child 1999;80:542-547.
- 239. Ramelli GP, Aloysius A, King C, Davis T, Muntoni F. Gastrostomy placement in paediatric patients with neuromuscular disorders: indications and outcome. Dev Med Child Neurol 2007;49:367-371.
Publication Dates
-
Publication in this collection
24 Mar 2009 -
Date of issue
Mar 2009
History
-
Accepted
17 Dec 2008 -
Received
31 Oct 2008