Abstracts
The congenital muscular dystrophies (CMDs) are a group of genetically and clinically heterogeneous hereditary myopathies with preferentially autosomal recessive inheritance, that are characterized by congenital hypotonia, delayed motor development and early onset of progressive muscle weakness associated with dystrophic pattern on muscle biopsy. The clinical course is broadly variable and can comprise the involvement of the brain and eyes. From 1994, a great development in the knowledge of the molecular basis has occurred and the classification of CMDs has to be continuously up dated. In the last number of this journal, we presented the main clinical and diagnostic data concerning the different subtypes of CMD. In this second part of the review, we analyse the main reports from the literature concerning the pathogenesis and the therapeutic perspectives of the most common subtypes of CMD: MDC1A with merosin deficiency, collagen VI related CMDs (Ullrich and Bethlem), CMDs with abnormal glycosylation of alpha-dystroglycan (Fukuyama CMD, Muscle-eye-brain disease, Walker Warburg syndrome, MDC1C, MDC1D), and rigid spine syndrome, another much rare subtype of CMDs not related with the dystrophin/glycoproteins/extracellular matrix complex.
congenital muscular dystrophy; MDC1A; collagen VI related disorders; glycosylation of alpha-dystroglycan; Fukuyama DMC; muscle-eye-brain (MEB) disease; Walker-Warburg syndrome; rigid spine syndrome
As distrofias musculares congênitas (DMCs) são miopatias hereditárias geralmente, porém não exclusivamente, de herança autossômica recessiva, que apresentam grande heterogeneidade genética e clínica. São caracterizadas por hipotonia muscular congênita, atraso do desenvolvimento motor e fraqueza muscular de início precoce associada a padrão distrófico na biópsia muscular. O quadro clínico, de gravidade variável, pode também incluir anormalidades oculares e do sistema nervoso central. A partir de 1994, os conhecimentos sobre genética e biologia molecular das DMCs progrediram rapidamente, sendo a classificação continuamente atualizada. Os aspectos clínicos e diagnósticos dos principais subtipos de DMC foram apresentados no número anterior deste periódico, como primeira parte desta revisão. Nesta segunda parte apresentaremos os principais mecanismos patogênicos e as perspectivas terapêuticas dos subtipos mais comuns de DMC: DMC tipo 1A com deficiência de merosina, DMCs relacionadas com alterações do colágeno VI (Ullrich e Bethlem), e DMCs com anormalidades de glicosilação da alfa-distroglicana (DMC Fukuyama, DMC "Muscle-eye-brain" ou MEB, síndrome de Walker Warburg, DMC tipo 1C, DMC tipo 1D). A DMC com espinha rígida, mais rara e não relacionada com alterações do complexo distrofina-glicoproteínas associadas-matriz extracelular também será abordada quanto aos mesmos aspectos patogênicos e terapêuticos.
distrofia muscular congênita; merosina; colágeno VI; glicosilação da alfa-distroglicana; DMC Fukuyama; DMC "muscle-eye-brain"-MEB; síndrome de Walker-Warburg; espinha rígida
VIEWS AND REVIEWS
Congenital muscular dystrophy. Part II: a review of pathogenesis and therapeutic perspectives
Distrofia muscular congênita. Parte II: revisão da patogênese e perspectivas terapêuticas
Umbertina Conti Reed
Departamento de Neurologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo SP, Brazil: Professora Titular da Disciplina de Neurologia Infantil
ABSTRACT
The congenital muscular dystrophies (CMDs) are a group of genetically and clinically heterogeneous hereditary myopathies with preferentially autosomal recessive inheritance, that are characterized by congenital hypotonia, delayed motor development and early onset of progressive muscle weakness associated with dystrophic pattern on muscle biopsy. The clinical course is broadly variable and can comprise the involvement of the brain and eyes. From 1994, a great development in the knowledge of the molecular basis has occurred and the classification of CMDs has to be continuously up dated. In the last number of this journal, we presented the main clinical and diagnostic data concerning the different subtypes of CMD. In this second part of the review, we analyse the main reports from the literature concerning the pathogenesis and the therapeutic perspectives of the most common subtypes of CMD: MDC1A with merosin deficiency, collagen VI related CMDs (Ullrich and Bethlem), CMDs with abnormal glycosylation of alpha-dystroglycan (Fukuyama CMD, Muscle-eye-brain disease, Walker Warburg syndrome, MDC1C, MDC1D), and rigid spine syndrome, another much rare subtype of CMDs not related with the dystrophin/glycoproteins/extracellular matrix complex.
Key words: congenital muscular dystrophy, MDC1A, collagen VI related disorders, glycosylation of alpha-dystroglycan, Fukuyama DMC, muscle-eye-brain (MEB) disease, Walker-Warburg syndrome, rigid spine syndrome.
RESUMO
As distrofias musculares congênitas (DMCs) são miopatias hereditárias geralmente, porém não exclusivamente, de herança autossômica recessiva, que apresentam grande heterogeneidade genética e clínica. São caracterizadas por hipotonia muscular congênita, atraso do desenvolvimento motor e fraqueza muscular de início precoce associada a padrão distrófico na biópsia muscular. O quadro clínico, de gravidade variável, pode também incluir anormalidades oculares e do sistema nervoso central. A partir de 1994, os conhecimentos sobre genética e biologia molecular das DMCs progrediram rapidamente, sendo a classificação continuamente atualizada. Os aspectos clínicos e diagnósticos dos principais subtipos de DMC foram apresentados no número anterior deste periódico, como primeira parte desta revisão. Nesta segunda parte apresentaremos os principais mecanismos patogênicos e as perspectivas terapêuticas dos subtipos mais comuns de DMC: DMC tipo 1A com deficiência de merosina, DMCs relacionadas com alterações do colágeno VI (Ullrich e Bethlem), e DMCs com anormalidades de glicosilação da alfa-distroglicana (DMC Fukuyama, DMC "Muscle-eye-brain" ou MEB, síndrome de Walker Warburg, DMC tipo 1C, DMC tipo 1D). A DMC com espinha rígida, mais rara e não relacionada com alterações do complexo distrofina-glicoproteínas associadas-matriz extracelular também será abordada quanto aos mesmos aspectos patogênicos e terapêuticos.
Palavras-chaves: distrofia muscular congênita, merosina, colágeno VI, glicosilação da alfa-distroglicana, DMC Fukuyama, DMC "muscle-eye-brain"-MEB, síndrome de Walker-Warburg, espinha rígida.
The congenital muscular dystrophies (CMDs) are genetically and clinically heterogeneous hereditary myopathies with a predominant autosomal recessive mode of inheritance that are characterized by congenital hypotonia, delayed motor development and early onset of progressive muscle weakness, as well as dystrophic pattern on muscle biopsy. The clinical course is broadly variable and can comprise the involvement of the brain and eyes1-7.
From 1994 and mostly in the first years of the current century a great input in the knowledge of the molecular basis has occurred, and the classification of CMDs has to be continuously up dated. The official journal of the World Muscular Society, Neuromuscular Disorders, periodically publishes the revised classification (Table 1)8. A computerized version of the classification is accessible at http://www.musclegenetable.org and http://194.167.35.195/. Most of the different genes involved with the pathogenesis of the CMD subtypes are related to the function of the dystrophin-glycoproteins associated complex (DGC) in the sarcolemma and extracellular matrix and their mutations lead either to defects in the glycosylation of alpha-dystroglycan (alpha-dystroglycanopathies) or to abnormalities of extracellular matrix proteins (MDC1A and collagen VI related disorders)1-7. A fourth subtype, rigid spine CMD, is related to a defect of an endoplasmic reticulum protein, selenoprotein N9, and recently a new subtype10 was associated to a defect of a nuclear protein, lamin A/C.
Last month, the first part of this review focused on the clinical and diagnostic aspects of the different subtypes of CMD. Presently, the second part emphasizes the main data on pathogenesis and therapeutic perspectives for the most common subtypes of CMD, i.e. MDC1A, collagen VI related disorders, CMDs caused by defects of glycosylation of alpha-DG, and finally the much rarer rigid spine CMD.
Before reviewing the essential pathogenic data about the most common subtypes of CMD, we summarize the general aspects of the organization of the DGC and extracellular matrix.
DYSTROPHIN-GLYCOPROTEINS ASSOCIATED COMPLEX AND EXTRACELLULAR MATRIX: GENERAL REMARKS ON STRUCTURE AND FUNCTION
The DGC is an assembly of proteins spanning the sarcolemma of skeletal muscle fibers that forms a chain of links between the contractile actin in the cytoskeleton and the extracellular matrix11,12 (Fig 1). Defects in the DGC can disrupt these links and alter the basement membrane organization, resulting in sarcolemmal instability and muscle cells apoptose. In addition to the function of stabilizing the sarcollema, the DCG is also essential in organizing molecules involved in cellular signaling12.
The first component of the chain of links in the inner cytoskeleton is dystrophin that through the amino-terminus domain binds to actin and through the carboxyl terminus binds to dystroglycan (DG)11,12. Dystrophin changes lead to the X-linked Duchenne and Becker muscular dystrophies, and are not involved in any type of CMD. The following link is dystroglycan (DG) that has two components: beta and alpha-DG. Beta-DG is a transmembrane glycoprotein and alpha-DG is extracellular but with close contact with the peripheral membrane11-13. Both are encoded by the same gene and then cleaved into two proteins, alpha and beta11-13. Primary mutations in the gene encoding DG have not been reported and the knockout mouse for DG is embryonically lethal14.
Alpha-DG is involved in the next step of the chain of links and connects the sarcolemma to the basement membrane. However, for performing this step, alpha-DG needs to be glycosylated11-15 (Fig 2). The glycosylation depends on the biosynthesis of glycans that occurs by the enzymatic activity of glycosyltransferases, and it is necessary for the correct function of many animal proteins, the glycoproteins. Dystroglycan glycosylation is highly conserved during evolution. The covalently addiction of sugar chains (glycans), forming a glycoprotein, induces a modification that increases the availability of the protein for ligand interaction with laminin, agrin and perlecan in skeletal muscle, as well as with laminin and neurexin in the brain11-18. The glycoproteins act as biosignals for cell-cell communication, intracellular signaling, protein folding, and targeting of proteins within cells11-15. Each glycosyltransferase has specific expression and localization in tissues or cell type as well as along the different stages of development, and its specificity is determined by the type of glycan component. The most common type of glycosylation that occurs in mammals' proteins is by N-glycan linkage. However, in a limited number of glycoproteins of brain, nerve, and skeletal muscle, including alpha-DG, the glycosylation is by O-glycan linkage and is named O-mannosyl-glycosylation because mannosylglycans are the specific sugars that promote the interaction between alpha-DG and extracellular matrix ligands. The glycosylation pattern of alpha-DG is specific not only for different tissues but also for different regions of the muscle fiber, such as the sarcolemma and the neuromuscular junction15. Protein O-mannose beta-1,2-N-acetylglucosaminyltransferase (POMGnT1) that was isolated in 200116, is the first human glycosyltransferase which was found to participate in O-mannosyl glycan synthesis in muscle and brain by adding N-acetylglucosamine to O-linked mannose16. Soon after, Michele et al.17 and Moore et al.18 suggested that, in addition to POMGnT1, fukutin and acetylglucosaminyl transferase-like protein (LARGE) may participate in a similar pathway "that ultimately results in transfers of sugar to dystroglycan". In patients with FCMD or MEB disease and in dystrophic mice they demonstrated that the O-glycosylation of alpha-DG, mediated by different glycosyltransferases, is essential for muscle and brain development and functions. During the O-linked mannose glycosilation the glycosyltransferases add different glycans directly to the hydroxyl groups of alpha-DG on either serine or threonine residues, following successive steps19. In this way, alpha-DG is sequentially modified by glycosyltransferases during the transportation from the rough endoplasmic reticulum to the trans-Golgi network20. Protein O-mannosyl tranferase 1 (POMT1) and protein O-mannosyl tranferase 2 (POMT2) act in endoplasmic reticulum during the first step of the O-mannosyl glycan synthesis21, and POMGnT1 acts during the second step in the Golgi apparatus16. POMT1 transfers a mannosyl residue from dolichyl phosphate mannose to a serine or threonine residue in alpha-DG, and POMGnT1 adds an N-acetylglucosamine residue16. Co-expression of POMT1 and POMT2 is necessary for O-mannosyl transferase activity within the ER of mammalian cells21. However, it is still unclear whether these glycans are directly involved in ligand binding or play some other role. Fukutin and FKRP are putative glycosyltransferases and their exact function and localization are controversial. Initially, it seemed that fukutin acted in the cis-Golgi compartment, while FKRP probably acted in the rough endoplasmic reticulum. Matsumoto et al.22 analysed the precise localization of fukutin and FKRP in muscle cells and suggested that FKRP localizes in the rough endoplasmic reticulum and with POMT1 may play a role in the first O-mannosylation step of α-DG. Others reported that FKRP and fukutin are localized subcellularly in the medial-Golgi apparatus23,24 but that the mutations in the FKRP gene would lead to endoplasmic reticulum retention and consequent diminution in the Golgi apparatus24. According to Yamamoto et al.25, although the functions of fukutin continue unclear, it seems to interact with alpha-DG during glycosylation but binding to the core area of alpha-DG instead of its sugar chain. Also concerning FKRP, it has recently been supposed that in normal and mutant mice it is detected in the sarcollema and coexists with DG in the native dystrophin-glycoprotein complex, perhaps having its localization mediated by DG26. It has not yet been demonstrated that LARGE is a glycosyltransferase itself but the Kanagawa et al.27 found that alpha-DG interacts with LARGE at two different domains, one involved with the glycosilation process itself and the other at intracellular level (N-terminal domain of α-DG) for defining a recognition motif necessary to initiate functional glycosylation. Therefore, in order to stimulate alpha-DG hyperglycosylation LARGE needs to both physically interact with alpha-DG intracellularly and function as a glycosyltransferase28. The two interactions are essential for the occurrence of the link between alpha-DG and laminin as well as other extracellular matrix ligands. LARGE also interacts with a homologous glycosyltransferase (glycosyltransferase-like 1B-GYLTL1B) named LARGE2 that is highly similar and also localizes to the Golgi apparatus but has lesser tissue expression and has not been detected in muscle and brain29. It is supposed that also LARGE2 is involved in alpha-DG glycosylation and that both show functional integration29. Although it has not yet been demonstrated that LARGE and LARGE 2 are glycosyltransferases themselves, their overexpression in cultured cells induce alpha-DG hyperglycosilation and laminin-binding; therefore, this biological activity in vivo suggests that both act as glycosyl transferases30. Indeed, LARGE2 was found to support the maturation of alpha-dystroglycan more effectively than LARGE31.
The structure, biosynthesis, and pathology of O-mannosyl glycans have been described in details by Endo13,15,32,33, Barresi and Campbell34 and Martin35. It is still under analyses whether these glycans are directly involved in ligand binding or play some other role30.
Molecular changes in genes that codified glycosyltransferases affecting this step of the DAG chain of links are responsible for the pathogenesis of five CMDs named alpha-dystroglycanopathies or CMDs by defects of the O-mannosyl-glycosylation of alpha-DG1-7,15,30,33,36-50: Fukuyama CMD (FCMD), "muscle-eye-brain" (MEB) disease, Walker-Warburg syndrome (WWS), MDC1C and MCD1D.
After being glycosylated alpha-DG links to the following component of the DAG chain, laminin alpha-2, a glycoprotein that together with collagen IV is a major constituent of the basement membrane of the extracellular matrix51 (Fig. 1). Its linkage to alpha-DG was firstly demonstrated by Ibraghimov-Beskrovnaya51 All the components of the laminin family are essential for the assembly and architectural integrity of the mammals' basement membranes52. The molecule of laminin is a heterotimer composed of three chains, alpha, beta e gamma, whose spatial arrangement has the form of a cross. The long arm of the cross corresponds to the long arms of the three chains closely assembled and each one of the three short arms of the cross comes from a different chain. At the moment 16 laminin isoforms are known, each one formed by a specific combination of alpha, beta, and gamma chain that allows a broad capacity of interaction with cellular receptors such as integrins and other extracellular ligands. Different laminins have their own spatial and temporal expression. In a recent review on the role of laminins in physiological and pathogenic conditions, Schéele et al.53 reported that one of the specific basement membrane functions is "to govern cell fate by inducing intracellular signalling cascades". In fact the broad spatial distribution of laminin genes products offers tissue specificity as each tissue has its proper ligands. The temporal expression of laminins influences the processes of proliferation, differentiation, adhesion, and migration in different stages of normal life or in pathologic states52. The role of laminin in the nervous system is less well defined than in the muscle, but its adhesive molecule is supposed to have an important role in peripheral nerve regeneration, influencing neurite outgrowth, neural differentiation, and synapse formation54.
Laminin-2 or laminin alpha-2, also named merosin, is a heterotrimer composed of alpha-2, beta-1, and gamma-1 chains, that is specifically found in the basement membranes of striated muscle, Schwann cells and placental trophoblasts55. Vuolteenaho et al.56 described the complete primary structure of the human laminin M chain (merosin), assigned it to 6q22-23 and reported its tecidual distribution, including in human fetal tissues. The term merosin was changed to muscle laminin alpha-2 by Burgeson et al.57 in 1994. In muscle, laminin alpha-2 acts as the most important ligand for the surface receptors of the muscular fiber, and is essential for controlling the transmission of force from the interior of the cytoskeleton. During the myogenesis, laminin alpha-2 is also involved in the stability and survival of the myotubes that develop normally in the first stages but collapsed and degenerated when the mature, contractile function of the muscular cell begins58. The biological functions of the rest of the chains of laminin complex (alpha-1, alpha-4 and alpha-5 chains) include the formation of basement membranes during muscle myogenesis and development, and probably a role in signal transmission events during muscle formation and muscle regeneration59.
Molecular changes in the gene responsible by codifying laminin alpha-2 are the source of the most common form of CMD, termed MDC1A or merosin-deficient CMD60.
In addition to the connection with alpha-DG, laminin alpha-2 also connects with the sarcolemma through the glycoprotein named integrin, and to the rest of the extracellular matrix network mainly through collagen IV unit (Fig 1).
Integrins are heterodimeric cell adhesion receptors organized in a transmembrane arrangement and extracellular domains that organize the cytoskeletand links to the extracellular matrix. They constitute the major cell surface receptors allowing cell-extracellular matrix adhesion61. In humans at least 18 alpha and eight beta subunits have been identified that compose 24 heterodimers with a great diversity of functions and tecidual specificity61,62. Playing as adhesion receptors, integrins act in the transduction of signals to the cell interior and receive intracellular signals that control their own ligand-binding affinity. Similarly to laminin, through these signaling pathways, integrins participate in proliferation, differentiation, apoptosis, and cell migration at different developmental stages61,62.
The specific integrin for the muscular fibre is integrin alpha-7/beta-1 that represents another link between the muscle fiber and the extracellular matrix that is independent of DG. Mutations in the integrin alpha7 gene are responsible by a very rare form of CMD63.
The link with laminin is the last step of the DAG chain but in the basement membrane, laminin alpha 2 binds to other laminins, nidogen (that binds to collagen IV and perlecan), agrin, fibronectin and different macromolecules64. In addition to laminin, type IV collagen is the most abundant glycoprotein in the basement membrane and all together the components of the basement membrane form a thin pericellular matrix that is involved in many cellular functions (gene expression, adhesion, migration, differentiation, and apoptosis)64,65. Through its carboxyl-terminal globular domain collagen IV interacts with the amino-terminal domain of the collagen VI alpha-1 chain65 and this connection is essential for anchoring the basement membrane of the muscular fiber to the other components of the extracellular matrix and maintaining the structural muscle integrity65-68. Collagen VI provides a microfilamentous network in the extracellular matrix of the muscular tissue, as well as in other organs66-68. Lampe and Bushby69 revised the literature concerning the main roles of collagen VI: it also interacts with several extracellular matrix components such as other fibrillar collagens, perlecan, glycoproteins associated to elastin, proteoglycans and fibronectin. In addition to these mechanical functions it also acts in cycle signaling, homeostasis control, tissue development and architecture, as well as in repair processes such as wound healing69.
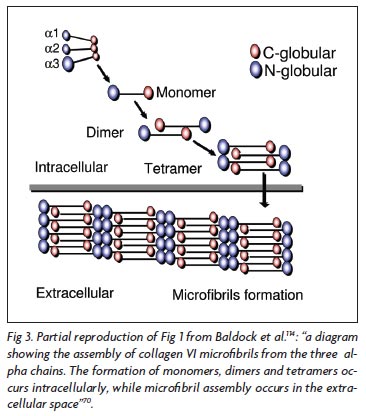
Collagen VI is formed by three chains: alpha-1, alpha-2, and alpha-3, each one encoded by a different gene. The structure and macromolecular organization of type VI collagen was described by Engel et al.66, and later by Baldock et al.70 and Ball et al.71 reported in details the biochemical basis of the tetrameric and microfibrillar organization. The three chains contribute to the formation of a triple helical monomer: first they associate through the C-terminal globular domain and then the triple helix folds from the C to N terminus to form the monomer that is the basic collagen VI structure66. The globular amino and carboxi-terminal regions of each monomer are assembled in an antiparallel manner by disulfide bonds into dimers that are aligned laterally to form tetramers. Each tetramer is threfore made by eight smaller subunits corresponding to the globular amino and carboxi-terminal regions of the monomeric components of the microfibril. The tetramers are then secreted and associated end-to-end to form a characteristic microfibrils repeat that constructs a network of microfibrils (Fig 3). The formation of monomers, dimers and tetramers occurs intracellularly, while the microfibril assembly occurs in the extracellular space66,69-71.
Mutations in the genes that codify the three collagen VI subunits lead to the second most common form of CMD, Ullrich atonic-sclerotic CMD72-75 and to Bethlem myopathy76,77 that now is included into CMD classification8.
After this brief explanation about the normal structure of the DAG/extracellular matrix complex we present the main pathogenic aspects and therapeutic perspectives of the most common forms of CMD that are caused by changes in this complex: CMD with merosin (laminin alpha-2) deficiency (MDC1A); collagen type VI related CMDs (Ullrich CMD and Bethlem myopathy); CMDs with abnormal glycosylation of alpha-DG (Fukuyama CMD, MEB disease, WWS, MDC1C, MDC1D).
Finally, we present the main pathogenic data and therapeutic researches about the much rare rigid spine CMD caused by a defect of an endoplasmic reticulum protein, selenoprotein N.
MEROSIN-DEFICIENT CONGENITAL MUSCULAR DYSTROPHY: MDC1A
Pathogenesis
MDC1A is caused by mutations in the laminin alpha-2 gene (LAMA2) and was first described by Tomé et al. in 199460. In the same year, Hillaire et al.78 demonstrated that this specific CMD is linked to 6q2, and soon after Heibling-Leclerc et al.79 found the first homozygous mutations in LAMA2 gene in two patients who had been reported by Tomé et al.60. The disease has congenital onset and progressive course and patients manifest severe muscular weakness and atrophy, diffuse contractures, inability to walk and facial dysmorphism61,2,60. Diffuse low-density areas in the cerebral white matter are the hallmark of this condition1,2,60. In a few patients who have partial laminin alpha-2 deficiency, a later onset and a milder course can be observed1,2. On muscle biopsies the dystrophic pattern is marked with fiber size variation, huge interstitial proliferation and fatty infiltration1,2,60. Necrosis and regeneration features are less prominent. Taniguchi et al.80 showed that regenerative features are less prominent in MDC1A and FCMD than in Duchenne muscular dystrophy. FCMD and MDC1A muscular samples of patients at different stages of life showed increased expression of extracellular matrix genes and downregulation of genes that codify structural components of mature muscle. This active interstitial fibrosis with less active regeneration of muscle cell components suggested that FCMD and MDC1A could be primary fibrotic diseases80.
Due to the fact that the alpha-2 subunit of laminin is also expressed in the basal lamina of Schwann cell-axon unit, a peripheral demyelinating neuropathy affecting predominantly motor fibers81, but also the sensitive ones82, is a feature of laminin alpha-2 -deficient CMD. In the dy mouse model laminin alpha-2 is expressed in the endoneurium surrounding the Schwann cell/myelin sheath and interacts with dystroglycan and/or alpha 6 beta 4 integrin located at the outer membrane of Schwann cell/myelin sheath. Its expression influences the peripheral myelinogenesis83. However, peripheral motor nerves involvement has not been found in Brazilian children with MDC 1A84,85 and this fact might be due to the type and location of the mutation84.
The characteristic pattern of white matter abnormality associated to MDC1A has been extensively analysed. Opposite to the peripheral nerve, in which laminin alpha 2 is associated only with myelinated and not with unmyelinated nerve fibers and is involved in the myelin stability86, a role for laminin alpha-2 in central myelination has not been confirmed. Villanova et al.87 found that laminin alpha-2 chain is localized to the basal lamina of all cerebral blood vessels and supposed that it may be important for the selective filtration capability of the blood-brain barrier. In patients with MDC1A the lack of laminin alpha-2 may lead to an abnormality of the blood-brain barrier causing impaired selective filtration. Caro et al.88 postulated that disruption of the blood-brain barrier associated with laminin alpha-2 leads to increased water content, resulting in abnormal white matter signal intensity. Using magnetic resonance spectroscopy and apparent diffusion coefficient mapping, Leite et al.89 detected abnormally high free-water concentrations in the white matter of our Brazilian patients with MDC1A and more prominent changes in the parietal, frontal, and temporal white matter. They also found no correlation between the extent of white matter abnormality on MRI and the clinical status as well as the degree of laminin alpha-2 deficiency (partial or total). Sijens et al.90 in a patient with MDC1A observed that the concentrations of all metabolites detected by brain magnetic resonance spectroscopy were decreased only in the white matter. This observation combined with fractional anisotropy decrease and apparent diffusion coefficient increase in the white matter indicated a presence of vasogenic edema in the white matter. The same was concluded by Brockmann et al.91 that observed a metabolite profile suggesting white matter edema which supports the hypothesis that laminin alpha2 deficiency results in leakage of fluids across the blood-brain barrier. Anyway, the pathophysiology of the white matter changes is not completely elucidated.
LAMA 2 mutations are markedly variable spanning all protein domains92-94 and, in general, the molecular diagnosis is not considered a priority in children with MDC1A, due to the homogeneous phenotype, the easy immunohistochemical analysis of laminin alpha-2 chain in muscle1,2,60 and skin95, and the characteristic white matter abnormalities on neuroimaging. However, Siala et al.96,97 have emphasized the utility of mRNA analysis in cases of MDC 1A to understand the mechanism of the mutation and the genotype-phenotype correlation. Recently, also Oliveira et al.98 emphasized the utility of analyzing genotype/phenotype correlations in MDC1A. In 26 patients they reported 96% of mutation detection rate using full genomic sequencing and complementary DNA analysis. They detected new polymorphisms and novel nonsense and truncating mutations, as well as the first fully characterized gross deletion. Interestingly, in the two patients with the milder clinical presentation and a partial reduction in muscle laminin-alpha2 one of the alleles showed a missense mutation that was not observed in any of the severe cases98.
Therapeutic perspectives
From 1994, knock-out mouse or spontaneous mutant mouse strains have been identified as animal models for MDC1A with total and partial deficiency99-105, and experimental therapeutic strategies have been attempted102,103,106-116. In mice, Kuang et al.102,103 were successful in obtaining the expression of a human laminin alpha 2 chain transgene under the regulation of a muscle-specific creatine kinase promoter. They detected the synthesis of laminin alpha-2 in muscle and a marked improvement of the clinical and histopathological changes; however, there was no improvement of the peripheral neuropathy and they concluded that potential new therapies have to be also directed to restore laminin alpha-2 in other tissue as CNS and peripheral nerves. Vilkin et al.106 studying both human and mouse myogenic cells verified that pure muscle cells are able to synthesize laminin alpha-2 chain. Using primary muscle cell culture transplantation, they found that laminin alpha-2 is secreted and expressed in both non-dystrophic muscles of an immunodeficient mouse and dystrophic muscles of a CMD animal model. However, not all myoblasts or muscle fibers that they form are capable of secretion and deposition in vivo as cell spreading and diffusion of the protein remain restricted to the injection site. In addition, the success of myoblast transplantation depends on future advances for controlling the immune response of the receptor and his biochemical adequacy with the donor. In dyW/dyW mice a "replacement therapy" using a mini-agrin (miniaturized form of agrin) resulted in a lesser muscle degeneration and in a decrease of mortality that is mediated through the linking of muscle basement membrane to the DGC, and not to integrin107. The first attempt of somatic gene therapy to treat mice with laminin alpha-2 deficiency was performed by Qiao et al.108. They used adeno-associated viral vector for delivering miniagrin gene into dyW/dyW mice and obtained an improvement of the dystrophic pattern. Mini-agrin not only ameliorates the phenotype in those mice but also decelerates disease progression when applied at late stages. Meinen et al.109 demonstrated that full-length agrin is also capable of promoting this improvement This result indicates the role of DG not only in the pathogenesis of CMDs caused by hypoglycosilation of alpha-DG, but also in MDC1A. The improvement of the phenotype may be due to the link between 'non-muscle' laminins and DG through miniagrin, therefore maintaining the sarcollema stability. These authors109 also supposed that since the genetic manipulation in dyW/dyW mice can prevent apoptosis, it might be possible that antitiapoptotic agents act synergistically with mini-agrin. Therefore, a future therapeutic attempt for patients with MDC1A could be the combination of antiapoptotic drugs with the expression of mini-agrin in muscle, and/or the up-regulation of endogenous agrin. Gawlik et al.110-112 generated laminin alpha-2 deficient mice expressing laminin alpha-1 in peripheral nerves and muscles. Dy3K/dy3K mice harboring the laminin alpha-1 transgene in these tissues presented reduced muscular dystrophy and show peripheral myelination. The finding that transgenically expressed laminin alpha-1compensates the lack of laminin alpha-2 in muscles and peripheral nerves indicates that laminin alpha-1 can be an ideal candidate for replacing laminin alpha-2 chain in MDC1A112. In addition, the authors considered that laminin alpha-1 overexpression induced by gene therapy would be paralogous, therefore minimizing adverse immunological responses. They also generated generated dy3k/dy3k laminin-a2 deficient mice in which the expression of alpha-DG in muscle is increased112. Using transgenic expression of laminin-a1, they obtained a restoration of the basement membrane and a normalization of alpha-DG hyperexpression, therefore demonstrating a probable interaction between laminin-a1 chain and alpha-DG112. Laminin-a1 also restored the link with the integrin at the sarcollema, what represents an additional way for increasing the muscle cell stability. Hagiwara et al.113 tested bone marrow transplantation in laminin-alpha2-deficient (dy) mice and reported an increase in lifespan, growth rate, muscle strength, and respiratory function. Xu et al.114 demonstrated that embryonic or postnatal overexpression of cytotoxic T cell GalNAc transferase (Galgt2) (the latter using adeno-associated virus) is effective in inhibiting the development of the muscular dystrophy or altering the disease progression in skeletal muscles of dy(w) by increasing the expression of agrin. Fukada et al.115 reported another possible therapeutic strategy for MDC1A. In normal animals, laminin alpha-2 is produced in CD90(+) cell fractions (from mesenchimal cells) not depending on its fusion with myogenic cells. They demonstrated that the number of C90+cells increases during muscle regeneration process in vivo and as those cells can be transplanted to the animal model of MDC 1A, perhaps they represent a new source of cellular theraphy. Allamand et al.116 using drugs that force reading through premature termination codons (PTCs) demonstrated that the mutant mRNAs were strongly stabilized in myotubes derived from MDC1 A after administration of negamycin but were not able to allow re-expression of laminin alpha-2, perhaps consequently to translational or post-translational troubles. They concluded that this novel form of therapy requires more studies to define the nucleotide context of PTCs, the mechanism of mRNA stability and its translation into a functional protein. Finally, the future treatment of lamininopathies may include stem-cell approaches as well as gene therapy53.
COLLAGEN VI RELATED MUSCLE DISORDERS
Pathogenesis
Mutations in each one of the three collagen VI genes, COL6A1 (21 q22.3), COL6A2 (21 q22. 3), and COL6A3 (2 q37) that encode respectively the alpha-1, alpha-2 and alpha-3 chains of collagen VI, cause two types of muscle disorders: Bethlem myopathy, with mild or moderate phenotype, and Ullrich CMD, with severe phenotype1-4,6,7,69,117. Bethlem myopathy is a slowly progressive disorder with variable age of onset, congenital or within the first or second decade of life, and marked flexion contractures of several joints7,69,76,77,117-119. Ullrich CMD is an early onset condition with severe weakness and progressive course that manifests proximal joint contractures and marked distal hyperlaxity. Protruding calcanei, follicular hyperkeratosis, and abnormal scarring are other frequent clinical signs1-4,6,7,69,72,73,118,119.
Until few years ago, Bethlem myopathy and Ullrich CMD were separate entities with distinct modes of inheritance; after the recognition of heterozygous in-frame deletions acting in a dominantly-negative way in patients with Ullrich phenotype the boundaries between Bethlem myopathy and Ullrich CMD became narrow69. According to Lampe and Bushby69 it is becoming clear that BM and UCMD represent opposite end points of a clinical continuum in which individuals presenting with intermediate phenotypes could be considered to have either ''mild UCMD'' or ''severe BM''. The concept of a spectrum of collagen VI-related disorders with marked clinical and genetic heterogeneity has emerged from the recent advances on the molecular mechanism of both diseases69. The complex genotype/phenotype correlations that have been broadly analysed in these two conditions clearly indicate that in both collagen VI-related disorders the main pathomechanism is due to the disruption of collagen VI anchorage to the basal lamina of the muscular fibers74,75,120-138.
Presently more than 60% of patients with collagen VI related disorders have autosomal dominant or recessive mutations identified121; however, the highly polymorphic nature of the three genes suggests the need of specific methods of mutations analysis for performing an adequate genetic counseling121. Since the triple helical deletions are readily detectable by RT-PCR analysis, Lampe et al.122 recommend that mutation analysis checks these regions first by RT-PCR prior to screen all the 107 coding exons.
The data from molecular analysis in Bethlem myopathy revealed that COL6A1 is the most involved gene and a splice site mutation seems to be the most common, not only in COL6A1 as also in COL6A2 and COL6A3 genes123,124. The a1exon 14 skipping mutation is the most commonly reported in Bethlem patients75,121,123-125, leading to reduced amounts of collagen VI protein, some of which is structurally abnormal with an impaired ability to form microfibrils69. As different kinds of mutations have been found, the pathogenic mechanism of Bethlem miopathy and the degree of clinical severity depend on the effect of the mutation on the structure, biosynthesis, secretion and assembly of collagen VI. In patients with different types of mutations, many have attempted to interpret the way by which the mutation alters the collagen VI organization and defines genotype/phenotype correlations. For example, a heterozygous nonsense mutation causing an alpha-1 premature stop codon and a decrease of available alpha-1 chains consequently reduces production and secretion of collagen VI126. Otherwise, a heterozygous missense mutation interrupting the repetitive amino acid sequence (glycine mutation) that forms the characteristic collagen triple helix either in the alpha-1or alpha-2 chains does not affect collagen VI intracellular monomer, dimer, and tetramer assembly or secretion, but the mutant tetramers are anomalous (kinked) and the extracellular microfibrils are functionally deficient127. Patients reported with a heterozygous in-frame deletion in the triple-helical domain resulting in exon skipping in the COL6A1 gene have either a typical clinical picture with finger contractures125 or a course more severe than that commonly reported128. Some data indicate that large deletions and mutations inside the triple-helical collagen VI monomer helix formed by alpha-1, 2 and 3 polypeptides are associated with a phenotype more severe than those mutations occurring in the amino-terminal globular region118. Pepe et al.129 found two large and highly similar heterozygous COL6A1 genomic deletions, spanning from intron 8 to exon 13 or intron 13, in two patients, one with Bethlem myopathy and the other with moderate Ullrich CMD. They noted that as a similar deletion had been previously reported in the severe Brazilian case with Ullrich CMD, the presence of a deletion-prone region involving the minisatellite in intron 8 of COL6A1 could be suggested129.
Lampe and Bushby69 summarized the most frequent pathogenic mechanisms in Bethlem myopathy: single amino acid substitutions disrupting the Gly-Xaa-Yaa motif of the triple helical domain in COL6A1, COL6A2, or COL6A377,121,127,130,and splice site mutations75,121,125,128 which cause skipping of COL6A1 exon 14 during pre-mRNA splicing and consequently in-frame deletion of 18 amino acids from the triple helical domain of the a1 chain. In the first case, mutations towards the N terminal region of the triple helix may cause kinking of the tetramers in the normally straight supercoiled triple helical region, thus reducing their ability to form microfibrils127 and exerting a dominant negative effect. Baker et al.124 confirmed that the majority of the Bethlem myopathy mutations occur toward the N-terminal end of the triple helix, that is a critical region of the collagen VI monomer (a1:a2:a3) is critical for efficient dimer, tetramer, and microfibril assembly. However, in the second case, the interpretation of the influence of missplicing mutations on collagen VI organization is difficult and needs caution69.
Recently, in one patient with Bethlem myopathy it was described a novel mutation that apparently did not affect the assembly, and it was suggested that its effect could be influencing collagen VI interactions in the extracellular matrix124.
In relation to Ullrich phenotype, the initial molecular studies in COL6A2 and COL6A3 genes pointed only to recessive mutations73,74,118. Lampe and Bushby69 revised the literature about the type of mutations in Ullrich CMD and reported a large number of the mutations that introduce premature termination codons with consequent nonsense mediated mRNA decay and loss of the mutated chain; another common type of mutation is by splicing that leads to in-frame exonic deletions as well as in-frame genomic deletions. Different missense changes can also occur within the triple helical and C terminal domains of COL6A2 and the N-terminal domains of COL6A3 their interpretation is difficult.
In 2003, the first heterozygous in-frame deletions acting in a dominantly-negative way was found in the COL6A1 gene75 in one Brazilian patient with severe Ullrich phenotype131; soon, more patients with a dominantly acting mutation in the COL6A1 were reported132-134, and finally this same type of mutation has also been found in Ullrich patients with mutations in COL6A2 and COL6A3 genes132. Heterozygously occurring N-terminal triple helical in-frame deletions allow mutant monomers to form dimers but secreted tetramers are abnormal with a consequent dominant negative effect on microfibrillar assembly132. In general, the complete absence of collagen VI in the extracellular matrix depends on mutations that exert a strong dominant-negative effect and compromise intracellular assembly of dimers, tetramers, and extracellular microfibrils132. However, the pathogenic effect of the in-frame heterozygous deletions is still poorly understood and the genetic counselling of patients and their families is a difficult task, as it has been also demonstrated by Peat et al.135.
Pace et al.136 recently considered that in spite of the great number of different types of mutations that have been already identified detailed analyses of the effects of the mutations on assembly of the protein have been conducted only in a small number of patients75,124,125,127,132. Pace et al.136 investigated in eight patients the mechanism by which different dominant glycine mutations in the collagen VI triple helix produce a spectrum of clinical phenotypes from mild Bethlem myopathy to severe UCMD, therefore pointing to the fact that the collagen VI related disorders form a continuum of clinical severity. Their study136 identified that in both, Bethlem and Ullrich CMD, heterozygous glycine mutations toward the N-terminal end of the triple helix impair the collagen VI assembly in two different ways, therefore producing different phenotypes: in the first that correspond to less compromised patients, collagen VI dimers accumulated in the cell but microfibril formation in the medium is moderately reduced, with no significant collagen VI decrease in the extracellular matrix; in the second, that corresponds to severely affected Ullrich patients, the assembly defects are more pronounced, some secreted collagen VI tetramers are not disulfide bonded, microfibril formation in the medium is altered, and collagen VI is decreased in the extracellular matrix136. Pace et al.136 supposed that the relative positions of the mutations contribute to understand why some glycine mutations in this region have no detectable effect on disulfide bonding whereas others result in secretion of nondisulfide bonded tetramers. In conclusion, they demonstrated that in patients with different dominant glycine mutations a mild phenotype is associated to a mild assembly defect whereas severe assembly defects result in moderate-to-severe phenotype.
Okada et al.138 found that in Japanese population mutations in collagen VI genes can lead to a collagen VI deficiency in the sarcollema (where normally it is strongly delineated) but with apparently normal amount in the interstitium. They sequenced the three collagen VI genes in 26 Japanese patients with primary collagen VI deficiency that in Japan accounts for 7.2% of CMD cases. By immunohistochemical analysis they found that most patients had sarcolemma-specific collagen VI deficiency and five had complete collagen VI deficiency, i.e. sarcollema plus interstitium. In the former group all mutations were sporadic dominant; however, in spite of the occurrence of this apparently specific localization of mutated collagen VI, they could not define any genotype/phenotype correlation. Kawahara et al.139,140 also found that in Ullrich CMD due to heterozygous mutations in COL6 genes collagen VI is preserved in the interstitium but lost in the sarcolemma. As in normal conditions collagen VI is particularly abundant close to the cells and in intimate contact with basement membranes surrounding myocytes, its lack caused by heterozygous mutations in COL6 genes diminish the anchorage of collagen VI microfibrils to the extracellular matrix.
Finally, Lampe et al.122 recently concluded that in collagen VI related disorders the genotype-phenotype correlation does exist and that the type and location of exon skipping mutation is predictive for severity and inheritance. They compared the molecular data of patients with Ullrich CMD with de novo dominant negative heterozygous splice mutations in COL6A1, COL6A2, and COL6A3, Ullrich CMD with recessively acting splice mutations, and Bethlem myopathy with heterozygous splice mutations. For the three types of mutations, i.e. dominant and recessive Ullrich CMD as well as dominant Bethlem myopathy, they discussed the different mechanisms by which the location of the skipped exon in relation to the molecular structure of the collagen chain and to the degree of interference on the collagen VI microfibrils assembly, strongly correlated with the clinical phenotype. Therefore, they add new evidence to the knowledge that in last instance the clinical severity depends on the ability of mutant chains to be incorporated in the final multimeric structure of collagen VI microfibrillar network122.
Although rarely, Ullrich phenotype can be detected even in patients who do not harbor mutations in the collagen VI genes and show a normal amount of collagen VI in the interstitium. Ishikawa et al.141 considered that these patients could have a primary abnormality of other not yet identified protein interacting with collagen VI in the sarcolemma causing a failure of collagen VI to anchor the basal lamina to the interstitium. However, according to Okada et al.138 the possibility of mutations affecting the promoter regions or introns, or of overlooked mutations must be considered in such cases.
In conclusion, it is clear that the complex genotype-phenotype correlations of collagen VI- related disorders are next to be elucidated. Recently137, three novel collagen VI chains, alpha-4, alpha-5, and alpha-6 were found in mice in or close to basement membrane. These chains structurally resemble the collagen VI alpha-3 chain, apparently having the ability of substitute it in the constitution of the heterotrimers. This identification could implicate in new findings related to the molecular biology of Bethlem myopathy and Ullrich CMD.
Therapeutic perspectives
As for other types of muscular dystrophies, many researches focusing on possible effective therapies have occurred. An animal model of human Bethlem myopathy has already been described and the details about the composition and the role of collagen VI have been widely discussed117,142.
For at least some types of mutations, Kawahara et al.139 suggested a therapeutic strategy based on a replacement therapy: in fibroblasts culture from patients with Ullrich CMD and COL6A1 glycine mutation, they verified that fibroblasts adhesion was markedly reduced but could be recovered by the addition of a medium with normal collagen VI139. Recently, Merlini et al.143 reported the results of an open pilot trial with oral cyclosporin A in five patients with collagen VI myopathies. After a month of treatment a new muscular biopsy showed a decrease of the mitochondrial and apoptotic changes in the myofibers as well as an increase of regenerative signs143. According to Angelin et al.144,145, an anomalous mitochondrial depolarization has been demonstrated in myoblasts from patients with Ullrich CMD, and cyclosporin A as well as intracellular Ca (2+) chelators, probably, prevent such anomalous mitochondrial depolarization and a consequent ATP depletion. Also Zou et al.146 recently reported another possible implication for future therapies. By means of immunofluorescence staining and Western blot analysis in vitro they found that the secretion and deposition of collagen VI in the extracellular matrix depends on interstitial fibroblasts but not on myogenic cells146. Studies on animal models for collagen VI related disorders may further offer good perspectives for therapeutic researches147,148.
DISORDERS OF GLYCOSYLATION OF ALPHA-DG (ALPHA-DYSTROGLYCANOPATHIES)
Pathogenesis
The alpha-dystroglycanopathies include different forms of dystrophies whose clinical severity varies from WWS that is associated with ocular abnormalities and CNS malformations and is not compatible with survival beyond the first years of life, to forms of LGMD with later onset later and pure muscular involvement. Five forms of CMDs are alpha-dystroglycanopathies1-7: FCMD, MEB disease,WWS, MDC1C and MDC1D (Table 2). FCMD, MEB disease and WWS form a spectrum of increasing clinical severity regarding muscle, eyes and brain malformation and MCD1D, with only one report, is also associated to severe muscular and CNS involvement. MDC1C can manifest pure, although severe, muscular involvement or presents associated clinical and radiological CNS involvement, suggested by mental retardation, cerebellar cysts and white matter abnormalities on neuroimaging149-151. The lack of CNS involvement in some patients can suggest that skeletal muscle is more sensitive to perturbation of dystroglycan30. The clinical and diagnostic particularities of each dystroglycanopathy were discussed in Part I of this review. The genetic and clinical heterogeneity of dystroglycanopathies are remarkable and, except for FCMD is impossible to correlate a particular gene with a specific phenotype (Table 2).
The pathogenic mechanism that explains the severe forms of CMDs associated with CNS manifestations emerged in 2001 from different works16, 152-154. Brockington et al.152 identified a gene that codifies a protein of the fukutin family termed fukutin related protein (FKRP) assigning it to 19q13.3. Mutations in the FKRP gene were found in children with a severe form of CMD who manifested inability to walk and muscle hypertrophy but no CNS changes and whose muscle samples showed a secondary deficiency of laminin alpha-2 immunostaining and a decreased expression of alpha-dystroglycan (DG). They named this new genotype/phenotype association as MDC1C and suggested that the mutations in the FKRP gene causing a defective glycosilation (hypoglycosilation) of alpha-DG would explain the basic pathologic mechanism in MDC1C152. Hayashi et al.153 found that the highly glycosylated epitope of alpha-DG was markedly deficient in skeletal and cardiac muscle and reduced in brain tissue obtained from patients with FCMD. These findings suggested that also fukutin could be involved in the glycosylation of alpha-DG that, when altered, could induce a disruption in the extracellular surface membrane of the muscle fiber, also influencing CNS development153. Yoshida et al.16 found that the MEB gene codifies a glycosyltransferase named POMGnT1 that is involved in O-mannosyl glycosylation and, therefore, suggested that an altered glycosylation could be a new pathomechanism for CMDs with neuronal migration disorder16. Kano et al.154 reported a selective deficiency of alpha-DG in MEB patients; therefore, they suggested that POMGNT1 acts on alpha-DG and that altered glycosylation of alpha-DG is involved in the pathogenesis of MEB.
At last, in 2002 two works from Campbells' research group at the Iowa University17,18 demonstrated that patients with MEB and FCMD, who carry mutant POMGnT1 and fukutin genes, respectively, had an abnormal hypoglycosilation of alpha-DG that abolished its binding with the ligands, laminin alpha-2, agrin, and neurexin of the extracellular matrix. The same occured in the myodystrophy (myd) mouse that carried a LARGE mutation and in the knock-out for brain-selective alpha-DG mouse. In the knock-out mouse they found aberrant cerebral and cerebellar migration probably derived from a discontinuos pial surface basal lamina (glia limitans); they considered that the aberrant migration, similar to that seen in patients with MEB and FCMD, was due to the lack of DG preventing its high-affinity binding to the extracellular matrix laminin. These results identify alpha-DG as having an essential role in both muscle and brain development and function and suggested that POMGnT1, fukutin, and LARGE may have a role in transferring sugars to alpha-DG, i.e. act as glycosyltransferases. The recognition of the involvement of other two glycosyltransferases, POMT1 and POMT2, in the pathogenesis of CMDs came soon after155-156.
During the last seven years, many have discussed and elucidated the pathogenesis of the defective glycosylation of alpha-DG, and the particular cortical involvement that is observed in many of them5,711,13,15,17,18,30,33,34,36-47,157-161. It is supposed that other subtypes of CMD will prove to depend on mutations in genes as yet unidentified but also involved in alpha-DG glycosylation39,45,46,48. In addition, from the first reports on congenital disorders of glycosylation, it seemed unlikely that the glycosyltransferases could have a role only in glycosylation of alpha-DG and the importance of looking for other protein targets has been emphasized37,38.
The role of alpha-DG in developing brain and in neuronal migration has been studied in animals models11,18,158, 159,162,163. Zaccaria et al.162 found that in adult mice alpha-DG is expressed in neurons of the cerebral cortex, hippocampus, olfactory bulb, basal ganglia, thalamus, hypothalamus, brainstem and cerebellum; in addition the astrocytes and their endfeet around blood vessels and the endothelial cells at the blood-brain barrier, also expressed DG. During CNS development, alpha-DG is expressed in the ventricular zone and in basement membranes, therefore participating in neuronal proliferation, in the constitution of the meningeal layer and in migration process as the radial glia is attached to the pial basement membrane158. In DG-null mice, then abnormal glycosylation of alpha-DG leads to different defects of neuronal migration that at the maximum of rupture of the pial-glial limitans, closely resemble the cobblestone aspect of cortex in human lissencephaly type II18.
The number of glycosiltransferases involved in the glycosylation of alpha-DG and their different possibilities of regional and developmental expression explain the spectrum of malformations variability and clinical severity that are observed in the different CMDs caused by alpha-dystroglycanopathies. Yamamoto et al.164 analysed the expression and localization of fukutin, POMGnT1, and POMT1 in the CNS and discussed the mechanism of lesions linked to these three glycosyltransferase whose changes are represented by migration disorders in FCMD, MEB, and WWS. They observed that fukutin, POMGnT1, and POMT1 are expressed especially in astrocytes that compose the astrocytic glia limitans and therefore is involved in the formation of the basement membrane. As the latter was found disrupted in fetal cases of FCMD, MEB, and WWS the defect in any of the three glycosyltransferases can be related to the pathogenesis of CNS lesions. These are also expressed in immature neurons what suggests their probable involvement in neuronal migration itself. In mature neurons POMGnT1 and POMT1 are expressed but fukutin is rarely positive indicating that at late stages of development POMGnT1 and POMT1 have a more crucial role than fukutin163. In addition, fukutin is expressed in pancreas, kidneys, bronchi, salivary gland, alimentary tract and skin in both fetal and adult mice, and in autopsied FCMD cases it was found decreased in kidneys, lung, skin and intestine163. These findings demonstrate the role of alpha-DG glycosylation also in non-muscle systems both during development and in the adult; studies with the aim of clarify why muscle and brain symptoms predominate in FCMD would be useful for better understanding the complex pathogenic mechanism of alpha-dystroglycanopathies163.
The wide phenotypical variation of the alpha-dystroglycanopathies, in particular the FKRP-related myopathies, has not a clear pathogenic basis. A study analysed patients with FKRP mutations who had also CNS alterations and concluded that the distribution of FKRP gene mutations did not allow any particular genotype/phenotype correlation concerning the amount of CNS malformations149.
Keramaris et al.24 considered that the type of missense point mutation could in part explain the phenotypical variation of FKRP gene mutations that alter the normal subcellular localization of FKRP protein in the Golgi apparatus: the protein is kept in the endoplasmic reticulum and diminishes in the Golgi apparatus. Therefore, each mutation could act in two ways, directly altering the presumed glycosyltransferase activity and further altering the protein function by the mislocalization24. In patients with FKRP mutations, the residual expression of alpha-DG detected by immunolabeling and Western blot can be correlated with the clinical severity and with the type of mutation168. Patients with MDC 1C who exhibit a severe phenotype are compound heterozygotes (one missense and one nonsense mutation) or have homozygous missense mutations; they present absent or strongly decreased α-DG expression. Patients with LGMD2I at the severe end of the clinical spectrum (Duchenne-like) tend to present a marked reduction in the immunolabeling of alpha-DG and are compound heterozygotes (a missense mutation and either another missense or a nonsense mutation) while patients at the milder end (later onset) generally present variable immunohistochemical pattern, sometimes only minimally changed, and in general are homozygous for the common C826A/Leu279Ile FKRP mutation168.
POMT1 and POMT2 mutations are not associated to the same clinical heterogeneity that is observed in the FKRP and in general cause classical WW or MEB phenotypes. However, out of these classical phenotypes, different types and degrees of cortical and posterior fossa malformations have been described156, 169-174, with or without eye involvement; mental retardation, and calf as well as thigh enlargement has also been related170,171. Yanagisawa et col172 hypothesized that patients with POMT1 and POMT2 mutations could share the same phenotype because, according to Manya et al.21, both glycosyltransferases form a heterodimeric complex that is responsible for the catalysis of the first step in O-mannosyl glycan synthesis. Messina et al.175 emphasized that POMT1 and POMT2 mutations have a wider clinical spectrum than initially thought and that, excluding MEB and WWS phenotypes, mutations causing frameshifts and stop codons were associated to the most severe phenotypes with predominant cerebellar hypoplasia commonly found. Godfrey et al.50 analysed eight patients with POMT1 mutations and nine with POMT2 mutations and suggested that POMT1 and POMT2 mutations do not seem to manifest a hierarchical involvement of muscle and brain, being associated with significant central nervous system involvement even in patients with relatively mild weaknessand who remain ambulant (LGMD2K). In this large revision about alpha-dystroglycanopathies not related to FKRP mutations Godfrey et al.50 found a broad correlation between the amount of depleted glycosylated epitope and phenotypic severity.
Recently another study165 demonstrated a good correlation between the clinical phenotype and the amount of glycosylated alpha-DG with the most severe cases being associated to a lack or marked reduction of glycosylated alpha-DG. However, this correlation was strong in patients with POMT1, POMT2 and POMGnT1 mutations, but not consistent in patients whose mutations had occurred in fukutin and FKRP genes.
The amount of dystroglycan hypoglycosylation is detected utilizing two monoclonal antibodies (IIH6 and VIA41) that recognize only the functional glycosylated epitope of alpha-DG30; further immunohistochemical studies will be useful for identifying all the glycan structures induced by glycosyltransferases, in particular LARGE activity30.
Considering all the alpha-dystroglycanopathies that have already been supposed by means of hypoglycosilation on the immunohistochemical analysis of muscle biopsies, Muntoni et al.5 pointed to a rate of confirmed molecular diagnosis around 65% of the cases, suggesting that more genes have yet to be identified. The same is clearly demonstrated by the work from Godfrey et al.50 that described many patients who had a phenotype suffesting alpha-dystroglycanopathy but no mutations in any of the six glycosyltransferases already identified. Therefore, they concluded that undefined, genes are likely to be involved in the pathogenesis of the dystroglycanopathies and that the identification of these genes may provide additional information on the pathway of glycosylation of alpha-DG.
Other great evidence pointing to the probability of the existence of more genes codifying glycosyltransferases not yet identified comes from the molecular studies from patients with WWS. In spite of its worldwide distribution and therefore the good availability of patients' DNA for genomic analysis, only one third of the cases has been associated to mutations in one of the six genes involved in the O-manosylation pathways50,166,167. It is supposed then that additional loci are going to be identified soon30,167.
Finally, Moore and Hewitt30 recently hypothesized that genetic variation between patients for other genes that might have compensatory functions could contribute to this clinical heterogeneity.
Therapeutic perspectives
Glycobiology of O-mannosyl glycans is a broad and open field of research that can favor the understanding of the pathogenesis and consequently the advent of a possible thearapeutic strategy for the alpha-dystroglycanopathies. The researches about the long-term potential of glycotherapies for CMD have begun with the first reports about the defective alpha-DG glycosylation as a new pathogenic mechanism for some subtypes of7,11,13,15,32-34,36-48,157,176,177. The central point of these researches is that the overexpression of LARGE promotes the attachement of glycans to alpha-DG, therefore restoring its ligand-binding function178.
Barresi et al.178 demonstrated that in Large(myd)mice overexpression of LARGE leads to a recovery of alpha-DG function as a receptor and ameliorates the dystrophic phenotype; in normal mice induces the synthesis of glycan-enriched alpha-DG with high affinity for extracellular ligands. They also demonstrated that in cell lines derived from patients with WWS and MEB (POMT1, POMGnT1 or fukutin mutations) as well as in transfected cell cultures from all cell lines, gene transfer of LARGE restores the laminin-binding activity of alpha-DG and results in hyperglycosylation of alpha-DG; these data indicate that, at least in this experimental over-expression system, LARGE may act in a different glycosylation pathway178. Therefore, up regulation of this gene might represent a therapeutic approach for the muscular dystrophies caused by defects of alpha-DG glycosyilation178. The Large myd mouse is an excellent and complete model for glycosylation-deficient CMDs as displays skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects179.
The LARGE homologue, glycosyltransferase-like 1B (GYLTL1B), named LARGE2, shows similar sequence and genomic organization and as LARGE-1 is localized in the Golgi apparatus, what suggests its function in glycosylation29. Since transient overexpression of LARGE2 in mouse myoblast cell line increased alpha-DG glycosylation and its affinity to bind laminin, Grewal et al.29 hypothesized that partial functional redundancy might occur between LARGE 1 and LARGE 2; therefore, the latter should be considered an additional potential therapeutic target. The details about the enzymatic activity of LARGE and LARGE 2 and the identification of the glycan structures that both induce remain undefined30. In addition, it is probable that LARGE 2 overexpression could compensate not only LARGE deficiency as also that of other glycosyltransferases177. Indeed, it is expected that LARGE 2 could be a candidate gene for an eventual non CMD-like genetic disease, as LARGE 2 has very low expression in muscle and brain30.
However, the CMDs caused by alpha-dystroglycanopathies constitute a therapeutic challenge because any intervention might be early, even prenatally and would need to reach multiple tissues. In addition to the potentially therapeutic overexpression of LARGE 1 and 2, other possible strategies have been discussed37,38,40,178,180,181: to obtain synthetic donors of sugars that could serve as glycan analogues; to carry on enzyme replacement via gene therapy, and to provide ectopic expression or upregulation of other glycosyltransferases that might compensate the loss of function of that altered by the mutation. A similar mechanism has been tested in a mouse model of Duchenne muscular dystrophy in which the dystrophic process was inhibited by the overexpression of Galgt2, another glycosyltransferase not implicated in CMD48,176,182. This promoted the synthesis of a form of a-DG that was followed by the recruitment of utrophin at the sarcollema, therefore restoring its stability. Recent works pointed to the fact that potential therapeutic strategies firstly depend on the complete elucidation of the molecular mechanism that is involved in the formation of DG structure, ligands and function, not only in muscles but also in non-neuromuscular tissues48,176.
The glycosylation itself is poorly understood and many points have to be considered. According to Sciandra et al.176, there might be that glycosyltransferases target other, unidentified, molecules that, hypoglycosylated, could also contribute to the pathogenesis of the alpha-dystroglycanopathies. They also stressed the importance of better analyzing the interaction between both, alpha and beta subunits, for DG function176.
Brancaccio157 discussed the possibility of the existence of a primary dystroglycanopathy, and hypoglycosylated, or even hyperglycosylated alpha-DG molecules also involved either in functional or pathological status; as the other authors above reported, he also emphasized that a possible therapeutic option could come from the compensatory glycosylation function of paralogue glycosyltransferases157.
Besides the therapeutic perspectives, in the topic correspondent to each subtype of CMD, we have already mentioned the genetic animal models, in general mice that have been studied in the researches on laminin alpha-2 deficient CMD, collagen VI related disorders, and many different types of alpha-dystroglycanopathies. Other, unusual, animal models have been utilized. In zebrafish, all the six identified or putative glycosyltransferases implicated in the pathogenic mechanism of the human muscular dystrophies were found to have similar patterns and expression than in patients, inclusively throughout early development. Therefore the zebrafish can be a useful model system for future researches183,184. Another experimental model for muscular dystrophies associated to disorders of alpha-DG glycosylation has also been found in mutant Drosophilas185 . The experimental studies in animal models provide new perspectives for clarifying the pathogenic mechanism of the CMDs and for testing potential therapeutic strategies. Unfortunately, until the moment the possibility of transferring some of the good results obtained in animal experiments to humans, in vivo, has to be considered with moderate optimism.
RIGID SPINE CMD
Pathogenesis
Rigid spine syndrome CMD is characterized by marked limitation in the flexion of the spine, gradual development of scoliosis leading to reduced respiratory vital capacity and respiratory failure as well as joints contractures. In 2001, Moghadaszadeh et al.9 identified the SEPN1 gene and several mutations resulting in rigid spine CMD, suggesting that selenoprotein N can be involved in redox reactions into the cell, protecting it from oxidant damage. Petit et al.186 demonstrated that SEPN1 is a glycoprotein-localized within the endoplasmic reticulum that is more abundant in human fetal tissues than in adult ones, including skeletal muscle. Although it is not a muscle specific protein and it is ubiquitously expressed, its expression is particularly marked in cultured myoblasts and decreases in differentiating myotubes, suggesting a role in early development and in cell proliferation and regeneration186. In zebrafish embryos deficient for SEPN, muscle organization and myofibrils architecture are altered during early development187.
In 2002, Ferreiro et al.188 concluded that rigid spine CMD and the most severe form of classic multiminicore disease are different phenotypes associated with mutations in SEPN1 gene. Later, they189 also demonstrated homozygous SEPN1 deletion in patients with CMD with Mallory body-like inclusions. Finally Clarke et al.190 described SEPN1mutations in two sisters with a diagnosis of typical congenital fiber-type disproportion without minicore lesions or dystrophic changes. Therefore the spectrum of selenoprotein-related disorders includes rigid spine CMD, multiminicore myopathy, desmin-related myopathy with Mallory body-like inclusions and congenital fiber-type disproportion. Rederstorff et al.191 confirmed the importance of selenium-containing proteins in muscle formation, maintenance and repair and attested that in patients with selenoprotein-related disorders most of mutations are nonsense mutations and deletions, likely to induce a loss of function.
As multiminicore myopathy is also caused by mutations in RyR1 gene that codifies the major component of the ryanodine receptor intracellular calcium release channel192,193, the relations ships between SEPN1 and RyR1 could explain this overlapping spectrum of congenital myopathies194. In the zebrafish embryo, Jurynec et al.194 demonstrated that the two proteins participate of the same cellular differentiation events and of the regulation of calcium fluxes. SEPN1 acts as a modifier of the RyR channel. In the absence of SEPN1, RyR channels lose their normal sensitivity to redox conditions. This finding explains because mutations affecting either factor lead to similar diseases194. Lescure et al.195 recently stressed the importance of also studying the correlation between SEPNI gene and protein and other genes involved in similar phenotypes, like desmin, alpha-actin and different selenoproteins acting in muscles and myocardium. In addition, the molecular function of SEPN and the role of the selenocystein (Sec) insertion that not only integrates the protein itself but also controls its mRNA quality needs to be further analysed195.
Therapeutic perspectives
Recently Rederstorff et al.196 reported a patient with a homozygous point mutation at the selenocystein (Sec) codon (c.G1385A) that converted the normal codon UGA to UAA, therefore preventing synthesis of a full-length active SePN protein. Then, they195 "engineered a mutant tRNASec gene carrying a point mutation in the anticodon, thereby restoring the base-pair complementarity with the SEPN1 mutated codon; in patient-derived primary fibroblasts the corrector tRNASec gene allowed read-through of the UAA stop codon, thus enabling synthesis of the full-length SePN protein"196. This is the first attempt of a therapeutic strategy for rigid spine CMD and other SEPN related disorders, using a mutation-specific approach. Considering the highly specific molecular context of SEPN1 mutations, the authors195 discuss in details and emphasize the efficiency and technical advantages of this alternative strategy in relation to conventional gene-therapy strategies.
In conclusion, the advances in the field of molecular genetics, cell biology and biochemistry have been the source of a better knowledge on the pathogenic mechanisms involved in the CMDs. However, the development of effective therapies is still at a preclinical stage, except for an attempt of treating patients with Ullrich phenotype with cyclosporin A143.
Unfortunately, the induction of a functional protein by gene modification, cell therapy or pharmacological agents has been a long and difficult task. The effective therapy is coming but employing it in large-scale clinical trials constitutes a challenge that is much greater in undeveloped countries.
Received 31 October 2008. Accepted 14 March 2009.
Dra. Umbertina Conti Reed - Avenida Dr. Enéas de Carvalho Aguiar 255/5º andar/sala 5131 - 05403-000 São Paulo SP - Brasil. E-mail: ucontireed@hcnet.usp.br
- 1. Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14:635-649.
- 2. Voit T, Tome FM. The congenital muscular dystrophies. Engel AG, Franzini-Armstrong C (Eds). Myology - basic and clinical. Edition 3. New York: McGraw-Hill-Medical Publishing Division, 2004:1203-1238.
- 3. Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F. Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci 2005;62:809-823.
- 4. Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006; 9:427- 443.
- 5. Muntoni F, Brockington M, Godfrey C, et al. Muscular dystrophies due to defective glycosylation of dystroglycan. Acta Myol 2007;26:129-135.
- 6. Lisi MT, Cohn RD. Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim Biophys Acta 2007;1772:159-172.
- 7. Schessl J, Zou Y, Bönnemann CG. Congenital muscular dystrophies and the extracellular matrix. Semin Pediatr Neurol 2006;13:80-89.
- 8. Gene table of monogenic neuromuscular disorders (nuclear genome only). Neuromuscular Disorders 2008;18:101-129.
- 9. Moghadaszadeh B, Petit N, Jaillard C, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet 2001; 29:17-18.
- 10. Quijano-Roy S, Mbieleu B, Bönnemann CG, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 2008; 64:177-186.
- 11. Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. Neuromuscul Disord 2005;15:207-217.
- 12. Ervasti JM, Sonnemann KJ. Biology of the striated muscle dystrophin-glycoprotein complex. Int Rev Cytol 2008; 265:191-225.
- 13. Endo T. O-mannosyl glycans in mammals. Biochim Biophys Acta 1999; 1473:237-246.
- 14. Williamson RA, Henry MD, Daniels KJ, et al. Dystroglycan is essential for early embryonic development: disruption of Reichert's; membrane in Dag 1-null mice. Hum Mol Genet 1997; 6:831-841.
- 15. Endo T, Toda T. Glycosylation in congenital muscular dystrophies. Biol Pharm Bull 2003; 26:1641-1647.
- 16. Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 2001; 1:717-724.
- 17. Michele DE, Barresi R, Kanagawa M, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002; 418:417-422.
- 18. Moore SA, Saito F, Chen J, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002; 418:422-425.
- 19. Chiba A, Matsumura K, Yamada H, et al. Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha-dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of alpha-dystroglycan with laminin. J Biol Chem 1997; 272:2156-2162.
- 20. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. Edition 4. New York: Garland Science, 2002.
- 21. Manya H, Chiba A, Yoshida A, et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA 2004; 101:500-505.
- 22. Matsumoto H, Noguchi S, Sugie K. Subcellular localization of fukutin and fukutin-related protein in muscle cells. J. Biochem 2004; 135: 709-712.
- 23. Esapa CT, Benson MA, Schroder JE, et al. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet 2002; 11:3319-3331.
- 24. Keramaris-Vrantsis E, Lu PJ, Doran T, et al. Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve 2007; 36:455-465.
- 25. Yamamoto T, Kawaguchi M, Sakayori N, et al. Intracellular binding of fukutin and alpha-dystroglycan: relation to glycosylation of alpha-dystroglycan. Neurosci Res 2006; 56:391-399.
- 26. Beedle AM, Nienaber PM, Campbell KP. Fukutin-related protein associates with the sarcolemmal dystrophin-glycoprotein complex. J Biol Chem 2007; 282:16713-16717.
- 27. Kanagawa M, Toda T. The genetic and molecular basis of muscular dystrophy: roles of cell-matrix linkage in the pathogenesis. J Hum Genet 2006; 51:915-926.
- 28. Brockington M, Torelli S, Prandini P, et al. Localization and functional analysis of the LARGE family of glycosyltransferases: significance for muscular dystrophy. Hum Mol Genet 2005; 14:657-665.
- 29. Grewal PK, McLaughlan JM, Moore CJ, Browning CA, Hewitt JE. Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 2005; 15:912-923.
- 30. Moore CJ, Hewitt JE. Dystroglycan glycosylation and muscular dystrophy. Glycoconj J. 2008 Sep 5. Epub ahead of print.
- 31. Fujimura K, Sawaki H, Sakai T, et al. LARGE2 facilitates the maturation of alpha-dystroglycan more effectively than LARGE. Biochem Biophys Res Commun 2005; 329:1162-1171.
- 32. Endo T. Structure, function and pathology of O-mannosyl glycans. Glycoconj J 2004; 21:3-7.
- 33. Endo T. Aberrant glycosylation of alpha-dystroglycan and congenital muscular dystrophies. Acta Myol 2005; 24:64-69.
- 34. Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci 2006;119:199-207.
- 35. Martin PT. Dystroglycan glycosylation and its role in matrix binding in skeletal muscle. Glycobiology 2005; 13:55-66.
- 36. Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet 2002; 360: 1419-1421.
- 37. Hewitt JE, Grewal PK. Glycosylation defects in inherited muscle disease. Cell Mol Life Sci 2003; 60:251-258.
- 38. Grewal PK, Hewitt JE. Glycosylation defects: a new mechanism for muscular dystrophy? Hum Mol Genet 2003; 12:259-264.
- 39. Martin PT, Freeze HH. Glycobiology of neuromuscular disorders. Glycobiology 2003; 13:67-75.
- 40. Martin-Rendon E, Blake DJ. Protein glycosylation in disease: new insights into the congenital muscular dystrophies. Trends Pharmacol Sci 2003; 24:178-183.
- 41. Muntoni F. Journey into muscular dystrophies caused by abnormal glycosylation. Acta Myol 2004; 23:79-84.
- 42. Muntoni F, Brockington M, Torelli S, Brown SC. Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol 2004; 17:205-209.
- 43. Schachter H, Vajsar J, Zhang W. The role of defective glycosylation in congenital muscular dystrophy. Glycoconj J 2004; 20:291-300.
- 44. Haliloglu G, Topaloglu H. Glycosylation defects in muscular dystrophies. Curr Opin Neurol 2004; 17:521-527.
- 45. Martin PT. The dystroglycanopathies: the new disorders of O-linked glycosylation. Semin Pediatr Neurol 2005;12:152-158.
- 46. Martin PT. Mechanisms of disease: congenital muscular dystrophies-glycosylation takes center stage. Nat Clin Pract Neurol 2006; 2:222-230.
- 47. Endo T, Manya H. Defect in glycosylation that causes muscular dystrophy. METHODs Enzymol 2006; 417:137-152.
- 48. Martin PT. Congenital muscular dystrophies involving the O-mannose pathway. Curr Mol Med 2007; 7:417-425.
- 49. Muntoni F, Brockington M, Godfrey C, et al. Muscular dystrophies due to defective glycosylation of dystroglycan. Acta Myol 2007;26:129-135.
- 50. Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007; 130:2725-2735.
- 51. Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 1992; 355:696-702.
- 52. Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol 2008;40:199-214.
- 53. Schéele S, Nyström A, Durbeej M, Talts JF, Ekblom M, Ekblom P. Laminin isoforms in development and disease. J Mol Med 2007; 85:825-836.
- 54. McGowan KA, Marinkovich MP. Laminins and human disease. Microsc Res Tech 2000; 51:262-279.
- 55. Leivo I, Engvall E. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA 1988; 85:1544-1548.
- 56. Vuolteenaho R, Nissinen M, Sainio K, et al. Human laminin M chain (merosin): complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. J Cell Biol 1994; 124:381-394.
- 57. Burgeson RE, Chiquet M, Deutzmann R, et al. A new nomenclature for the laminins. Matrix Biol 1994; 14:209-211.
- 58. Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E. Merosin and laminin in myogenesis; specific requirement for merosin in myotube stability and survival. J Cell Biol 1996; 134:1483-1497.
- 59. Gullberg D, Tiger CF, Velling T. Laminins during muscle development and in muscular dystrophies. Cell Mol Life Sci 1999;56:442-460.
- 60. Tome FM, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci 1994;III 317; 351-357.
- 61. Takagi J. Structural basis for ligand recognition by integrins. Curr Opin Cell Biol 2007; 19:557-564.
- 62. Takada Y, Ye X, Simon S. The integrins. Genome Biol 2007; 8:215-223.
- 63. Hayashi YK, ChouFL, Engvall E, et al. Mutations in the integrin alpha-7 gene cause congenital myopathy. Nature Genet 1998;19:94-97.
- 64. Sanes JR. The basement membrane/basal lamina of skeletal muscle. J Biol Chem 2003; 278:12601-12604.
- 65. Kuo HJ, Maslen CL, Keene DR, Glanville RW. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J Biol Chem 1997; 272:26522-26529.
- 66. Engel J, Furthmayr H, Odermatt E, et al. Structure and macromolecular organization of type VI collagen. Ann N Y Acad Sci 1985; 460:25-37.
- 67. Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004;131:1619-1628.
- 68. LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med (Maywood) 2007; 232:1121-1129.
- 69. Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet 2005; 42:673-685.
- 70. Baldock C, Sherratt MJ, Shuttleworth CA, Kielty CM. The supramolecular organization of collagen VI microfibrils. J Mol Biol 2003; 330:297-307.
- 71. Ball S, Bella J, Kielty C, Shuttleworth A. Structural basis of type VI collagen dimer formation. J Biol Chem 2003;278:15326-15332.
- 72. Ullrich O. Kongenitale atonisch-sklerotische Muskeldystrophie, ein weiterer Typus der heredodegeneration Erkrankungen des nueromuskularen Systems. Z Ges Neurol Psychiat 1930; 126:171-201.
- 73. Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA 2001; 98:7516-7521.
- 74. Demir E, Sabatelli P, Allamand V, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet 2002; 70:1446-1458.
- 75. Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet 2003; 73:355-369.
- 76. Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain 1976;99:91-100.
- 77. Jobsis GJ, Keizers H, Vreijling JP, et al. Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat Genet 1996;14:113-115.
- 78. Hillaire D, Leclerc A, Faure S, et al. Localization of merosin-negative congenital muscular dystrophy to chromosome 6q2 by homozygosity mapping. Hum Molec Genet 1994;3:1657-1661.
- 79. Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin alpha-2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nature Genet 1995;11:216-218.
- 80. Taniguchi M, Kurahashi H, Noguchi S, et al. Expression profiling of muscles from Fukuyama-type congenital muscular dystrophy and laminin-alpha 2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem Biophys Res Commun 2006; 342:489-502.
- 81. Shorer Z, Philpot J, Muntoni F, Sewry C, Dubowitz V. Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J. Child Neurol 1995;10:472-475.
- 82. Quijano-Roy S, Renault F, Romero N, Guicheney P, Fardeau M, Estournet B. EMG and nerve conduction studies in children with congenital muscular dystrophy. Muscle Nerve 2004; 29:292-299.
- 83. Matsumura K, Yamada H, Saito F, Sunada Y, Shimizu T. Peripheral nerve involvement in merosin-deficient congenital muscular dystrophy and dy mouse. Neuromuscul Disord 1997; 7:7-12.
- 84. Ferreira LG, Marie SK, Liu EC, et al. Dystrophin-glycoproteins associated in congenital muscular dystrophy: immunohistochemical analysis of 59 Brazilian cases. Arq Neuropsiquiatr 2005; 63:791-800.
- 85. Reed UC, Marie SK, Vainzof M, et al. Congenital muscular dystrophy with cerebral white matter hypodensity. Correlation of clinical features and merosin deficiency. Brain Dev 1996;18:53-58.
- 86. Malandrini A, Villanova M, Sabatelli P, et al. Localization of the laminin alpha 2 chain in normal human skeletal muscle and peripheral nerve: an ultrastructural immunolabeling study. Acta Neuropathol 1997; 93:166-172.
- 87. Villanova M, Malandrini A, Sabatelli P, et al. Localization of laminin alpha 2 chain in normal human central nervous system: an immunofluorescence and ultrastructural study. Acta Neuropathol 1997; 94:567-571.
- 88. Caro PA, Scavina M, Hoffman E, Pegoraro E, Marks HG. MR imaging findings in children with merosin-deficient congenital muscular dystrophy. AJNR Am J Neuroradiol 1999; 20:324-326.
- 89. Leite CC, Reed UC, Otaduy MC, et al. Congenital muscular dystrophy with merosin deficiency: 1H MR spectroscopy and diffusion-weighted MR imaging. Radiology 2005; 235:190-196.
- 90. Sijens PE, Fock JM, Meiners LC, Potze JH, Irwan R, Oudkerk M. MR spectroscopy and diffusion tensor imaging of the brain in congenital muscular dystrophy with merosin deficiency: metabolite level decreases, fractional anisotropy decreases, and apparent diffusion coefficient increases in the white matter. Brain Dev 2007; 29:317-321.
- 91. Brockmann K, Dechent P, Bönnemann C, Schreiber G, Frahm J, Hanefeld F. Quantitative proton MRS of cerebral metabolites in laminin alpha2 chain deficiency. Brain Dev 2007; 29:357-364.
- 92. Di Blasi C, Piga D, Brioschi P, et al. LAMA2 gene analysis in congenital muscular dystrophy: new mutations, prenatal diagnosis, and founder effect. Arch Neurol 2005; 62:1582-1586.
- 93. Tezak Z, Prandini P, Boscaro M, et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha-2 (LAMA2) deficiency. Hum Mutat 2003; 21:103-111.
- 94. Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: genotype/phenotype studies of 22 patients. Neurology 1998; 51:101-110.
- 95. Sewry CA, D'Alessandro M, Wilson LA, et al. Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics 1997; 28:217-222.
- 96. Siala O, Louhichi N, Triki C, et al. Severe MDC1A congenital muscular dystrophy due to a splicing mutation in the LAMA2 gene resulting in exon skipping and significant decrease of mRNA level. Genet Test 2007; 11:199-207.
- 97. Siala O, Louhichi N, Triki C, Morinière M, Fakhfakh F, Baklouti F. LAMA2 mRNA processing alterations generate a complete deficiency of laminin-alpha2 protein and a severe congenital muscular dystrophy. Neuromuscul Disord 2008; 18:137-145.
- 98. Oliveira J, Santos R, Soares-Silva I, et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet 2008 Aug 12. Epub ahead of print.
- 99. Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J Biol Chem 1994; 269:13729-13732.
- 100. Xu H, Christmas P, Wu XR, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Nat Acad Sci 1994; 91:5572-5576.
- 101. Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha-2 (Lama2) gene. Nature Genet 1994; 8:297-302.
- 102. Kuang W, Xu H, Vachon PH, Engvall E. Disruption of the lama2 gene in embryonic stem cells: laminin alpha 2 is necessary for sustenance of mature muscle cells. Exp Cell Res 1998; 241:117-125.
- 103. Kuang W, Xu H, Vachon P, et al. Merosin-deficient congenital muscular dystrophy: partial genetic correction in two mouse models. J. Clin Invest 1998; 102:844-852.
- 104. Guo LT, Zhang XU, Kuang W, et al. Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul Disord 2003; 13:207-215.
- 105. Miyagoe Y, Hanaoka K, Nonaka I, et al. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett 1997; 415:33-39.
- 106. Vilquin JT, Guérette B, Puymirat J, et al. Myoblast transplantations lead to the expression of the laminin alpha 2 chain in normal and dystrophic (dy/dy) mouse muscles. Gene Ther 1999; 6:792-800.
- 107. Moll J, Barzaghi P, Lin S, et al. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 2001; 413:302-307.
- 108. Qiao C, Li J, Zhu T, et al. Amelioration of laminin-2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA 2005;102:11999-12004.
- 109. Meinen S, Barzaghi P, Lin S, Lochmüller H, Ruegg MA. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J Cell Biol 2007; 176:979-993.
- 110. Gawlik K, Miyagoe-Suzuki Y, Ekblom P, Takeda S, Durbeej M. Laminin a1 chain reduces muscular dystrophy in laminin a2 chain deficient mice. Hum Mol Genet 2004; 13:1775-1784.
- 111. Gawlik KI, Li JY, Petersén A, Durbeej M. Laminin a1-chain improves laminin a2-chain deficient peripheral neuropathy. Hum Mol Genet 2006; 15:2690-2700.
- 112. Gawlik KI, Mayer U, Blomberg K, Sonnenberg A, Ekblom P, Durbeej M. Laminin a1-chain-mediated reduction of laminin a2-chain-deficient muscular dystrophy involves integrin a7b1 and dystroglycan. FEBS Lett 2006; 580:1759-1765.
- 113. Hagiwara H, Ohsawa Y, Asakura S, Murakami T, Teshima T, Sunada Y. Bone marrow transplantation improves outcome in a mouse model of congenital muscular dystrophy. FEBS Lett 2006; 580:4463-4468.
- 114. Xu R, Chandrasekharan K, Yoon JH, Camboni M, Martin PT. Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am J Pathol 2007; 171:181-199.
- 115. Fukada S, Yamamoto Y, Segawa M, et al. CD90-positive cells, an additional cell population, produce laminin alpha2 upon transplantation to dy(3k)/dy(3k) mice. Exp Cell Res 2008; 314:193-203.
- 116. Allamand V, Bidou L, Arakawa M, et al. Drug-induced readthrough of premature stop codons leads to the stabilization of laminin alpha2 chain mRNA in CMD myotubes. J Gene Med 2008; 10:217-224.
- 117. Pepe G, de Visser M, Bertini E, et al. Bethlem myopathy (BETHLEM) 86th ENMC international workshop, 10-11 November 2000, Naarden, The Netherlands. Neuromuscul Disord 2002; 12:296-305.
- 118. Pepe G, Bertini E, Bonaldo P, et al. Bethlem myopathy (BETHLEM) and Ullrich scleroatonic muscular dystrophy: 100th ENMC international workshop, 23-24 November 2001, Naarden, The Netherlands. Neuromuscul Disord 2002; 12:984-993.
- 119. Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol 2002; 6:193-198.
- 120. Demir E, Ferreiro A, Sabatelli P, et al. Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics 2004; 35:103-112.
- 121. Lampe AK, Dunn DM, von Niederhausern AC, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet 2005; 42:108-120.
- 122. Lampe AK, Zou Y, Sudano D, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat 2008; 29:809-822.
- 123. Lucioli S, Giusti B, Mercuri E, et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology 2005; 64:1931-1937.
- 124. Baker NL, Mörgelin M, Pace RA, et al. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol 2007; 62:390-405.
- 125. Lamande SR, Shields KA, Kornberg AJ, Shield LK, Bateman JF. Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem 1999; 274:21817-21822.
- 126. Lamande SR, Bateman JF, HutchisonW, et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet 1998; 7:981-989.
- 127. Lamande SR, Morgelin M, Selan C, Jobsis GJ, Baas F, Bateman JF. Kinked collagen VI tetramers and reduced microfibril formation as a result of Bethlem myopathy and introduced triple helical glycine mutations. J Biol Chem 2002; 277:1949-1956.
- 128. Pepe G, Giusti B, Bertini E, et al. A heterozygous splice site mutation in COL6A1 leading to an in-frame deletion of the alpha1(VI) collagen chain in an Italian family affected by Bethlem myopathy. Biochem Biophys Res Commun 1999; 258:802-807.
- 129. Pepe G, Lucarini L, Zhang RZ, et al. COL6A1 genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy. Ann Neurol 2006; 59:190-195.
- 130. Pepe G, Bertini E, Giusti B, et al. A novel de novo mutation in the triple helix of the COL6A3 gene in a two-generation Italian family affected by Bethlem A diagnostic approach in the mutations' screening of type VI collagen myopathy. Neuromuscul Disord 1999; 9:264-271.
- 131. Reed UC, Ferreira LG, Liu EC, et al. Ullrich congenital muscular dystrophy and Bethlem myopathy: clinical and genetic heterogeneity. Arq Neuropsiquiatr 2005; 63:785-790.
- 132. Baker NL, Morgelin M, Peat R, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 2005; 14:279-293.
- 133. Jimenez-Mallebrera C, Maioli MA, Kim J, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul Disord 2006; 16:571-582.
- 134. Giusti B, Lucarini L, Pietroni V, et al. Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol 2005;58:400-410.
- 135. Peat RA, Baker NL, Jones KJ, North KN, Lamandé SR. Variable penetrance of COL6A1 null mutations: implications for prenatal diagnosis and genetic counselling in Ullrich congenital muscular dystrophy families. Neuromuscul Disord 2007; 17:547-557.
- 136. Pace RA, Peat RA, Baker NL, et al. Collagen VI glycine mutations: perturbed assembly and a spectrum of clinical severity. Ann Neurol 2008; 64:294-303.
- 137. Gara SK, Grumati P, Urciuolo A, et al. Three novel collagen VI chains with high homology to the alpha3 chain. J Biol Chem 2008; 283:10658-10670.
- 138. Okada M, Kawahara G, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007; 69:1035-1042.
- 139. Kawahara G, Okada M, Morone N, et al. Reduced cell anchorage may cause sarcolemma-specific collagen VI deficiency in Ullrich disease. Neurology 2007; 69:1043-1049.
- 140. Kawahara G, Ogawa M, Okada M, et al. Diminished binding of mutated collagen VI to the extracellular matrix surrounding myocytes. Muscle Nerve 2008; 38:1192-1195.
- 141. Ishikawa H, Sugie K, Murayama K, et al. Ullrich disease due to deficiency of collagen VI in the sarcolemma. Neurology 2004; 62:620-623.
- 142. Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet 1998; 7:2135-2140.
- 143. Merlini L, Angelin A, Tiepolo T, et al. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc Natl Acad Sci USA 2008; 105:5225-5229.
- 144. Angelin A, Tiepolo T, Sabatelli P, et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci USA 2007; 104:991-996.
- 145. Angelin A, Bonaldo P, Bernardi P. Altered threshold of the mitochondrial permeability transition pore in Ullrich congenital muscular dystrophy. Biochim Biophys Acta 2008; 1777:893-896.
- 146. Zou Y, Zhang RZ, Sabatelli P, Chu ML, Bönnemann CG. Muscle interstitial fibroblasts are the main source of collagen VI synthesis in skeletal muscle: implications for congenital muscular dystrophy types Ullrich and Bethlem. J Neuropathol Exp Neurol 2008; 67:144-154.
- 147. Irwin WA, Bergamin N, Sabatelli P, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 2003; 35:367-371.
- 148. Myllyharju J, Kivirikko KI. Collagens and collagen-related diseases. Ann Med 2001; 33:7-21.
- 149. Mercuri E, Topaloglu H, Brockington M, et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol 2006; 63:251-257.
- 150. Louhichi N, Triki C, Quijano-Roy S, et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 2004; 5:27-34.
- 151. Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 2003 60:988-992.
- 152. Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha-2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001; 69:1198-1209.
- 153. Hayashi YK, Ogawa M, Tagawa K, et al. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001; 57:115-121.
- 154. Kano H, Kobayashi K, Herrmann R, et al. Deficiency of alpha-dystroglycan in muscle-eye-brain disease. Biochem Biophys Res Commun 2002; 291:1283-1286.
- 155. Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002;71:1033-1043.
- 156. van Reeuwijk J, Janssen M, van den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 2005; 42:907-912.
- 157. Brancaccio A. Alpha-dystroglycan, the usual suspect? Neuromuscul Disord 2005; 15:825-828.
- 158. Montanaro F, Carbonetto S. Targeting dystroglycan in the brain. Neuron 2003; 37:193-196.
- 159. Qu Q, Smith FI. Alpha-dystroglycan interactions affect cerebellar granule neuron migration. J Neurosci Res 2004; 76:771-782.
- 160. Saito Y, Kobayashi M, Itoh M, et al. Aberrant neuronal migration in the brainstem of Fukuyama-type congenital muscular dystrophy. J Neuropathol Exp Neurol 2003; 62:497-508.
- 161. Saito Y, Yamamoto T, Mizuguchi M, et al. Altered glycosylation of alpha-dystroglycan in neurons of Fukuyama congenital muscular dystrophy brains. Brain Res 2006;1075:223-228.
- 162. Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience 2001; 104:311-24.
- 163. Saito Y, Yamamoto T, Ohtsuka-Tsurumi E, et al. Fukutin expression in mouse non-muscle somatic organs: its relationship to the hypoglycosylation of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Brain Dev 2004; 26:469-479.
- 164. Yamamoto T, Kato Y, Kawaguchi M, Shibata N, Kobayashi M. Expression and localization of fukutin, POMGnT1, and POMT1 in the central nervous system: consideration for functions of fukutin. Med Electron Microsc 2004; 37:200-207.
- 165. Jimenez-Mallebrera C, Torelli S, Feng L, et al. A Comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 2008; Aug 7. Epub ahead of print.
- 166. Cotarelo RP, Valero MC, Prados B, et al. Two new patients bearing mutations in the fukutin gene confirm the relevance of this gene in Walker-Warburg syndrome. Clin Genet 2008; 73:139-145.
- 167. van Reeuwijk J, Brunner HG, van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet 2005;67:281-289.
- 168. Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol 2004; 164:727-737.
- 169. Balci B, Uyanik G, Dincer P, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord 2005; 15:271-275.
- 170. van Reeuwijk J, Maugenre S, van den Elzen C, et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat 2006; 27:453-459.
- 171. Mercuri E, D'Amico A, Tessa A, et al. POMT2 mutation in a patient with 'MEB-like' phenotype. Neuromuscul Disord 2006; 16:446-448.
- 172. Yanagisawa A, Bouchet C, Van den Bergh PY, et al. New POMT2 mutations causing congenital muscular dystrophy: identification of a founder mutation. Neurology 2007;69:1254-1260.
- 173. Dincer P, Balci B, Yuva Y, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of alpha-dystroglycan. Neuromuscul Disord 2003;13:771-778.
- 174. D'Amico A, Tessa A, Bruno C, et al. Expanding the clinical spectrum of POMT1 phenotype. Neurology 2006; 66:1564-1567.
- 175. Messina S, Mora M, Pegoraro E, et al. POMT1 and POMT2 mutations in CMD patients: a multicentric Italian study. Neuromuscul Disord 2008; 18:565-571.
- 176. Sciandra F, Gawlik KI, Brancaccio A, Durbeej M. Dystroglycan: a possible mediator for reducing congenital muscular dystrophy? Trends Biotechnol 2007; 25:262-268.
- 177. Brockington M, Muntoni F. The modulation of skeletal muscle glycosylation as a potential therapeutic intervention in muscular dystrophies. Acta Myol 2005; 24:217-221.
- 178. Barresi R, Michele DE, Kanagawa M, et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies.Nat Med 2004; 10:696-703.
- 179. Holzfeind PJ, Grewal PK, Reitsamer HA., et al. Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Largemyd mouse defines a natural model for glycosylation-deficient muscle-eye-brain disorders. Hum Mol Genet 2002; 11:2673-2687.
- 180. Muntoni F, Brockington M, Brown SC. Glycosylation eases muscular dystrophy. Nat Med 2004;10:676-677.
- 181. Rando TA. Artificial sweeteners-enhancing glycosylation to treat muscular dystrophies. N Engl J Med 2004; 35:1254-1256.
- 182. Nguyen HH, Jayasinha V, Xia B, Hoyte K, Martin PT. Overexpression of cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci USA 2002; 99:5616-5621.
- 183. Moore CJ, Goh HT, Hewitt JE. Genes required for functional glycosylation of dystroglycan are conserved in zebrafish. Genomics 2008; 92:159-167.
- 184. Thornhill P, Bassett D, Lochmüller H, Bushby K, Straub V. Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP). Brain 2008;131:1551-1561.
- 185. Wairkar YP, Fradkin LG, Noordermeer JN, DiAntonio A. Synaptic defects in a Drosophila model of congenital muscular dystrophy. J Neurosci 2008; 28:3781-3789.
- 186. Petit N, Lescure A, Rederstorff M, et al. Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet 2003; 12:1045-1053.
- 187. Deniziak M, Thisse C, Rederstorff M, Hindelang C, Thisse B, Lescure A.. Loss of selenoprotein N function causes disruption of muscle architecture in the zebrafish embryo. Exp Cell Res 2007; 313:156-167.
- 188. Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002; 71:739-749.
- 189. Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol 2004; 55:676-686.
- 190. Clarke NF, Kidson W, Quijano-Roy S, et al.SEPN1: associated with congenital fiber-type disproportion and insulin resistance. Ann Neurol 2006; 59:546-552.
- 191. Rederstorff M, Krol A, Lescure A. Understanding the importance of selenium and selenoproteins in muscle function.Cell Mol Life Sci 2006; 63:52-59.
- 192. Monnier N, Ferreiro A, Marty I, Labarre-Vila A, Mezin P, Lunardi J. A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet 2003; 12:1171-1178.
- 193. Jungbluth H, Zhou H, Hartley L, et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology 2005; 65:1930-1935.
- 194. Jurynec MJ, Xia R, Mackrill JJ, et al. Selenoprotein N is required for ryanodine receptor calcium release channel activity in human and zebrafish muscle. Proc Natl Acad Sci U S A. 2008; 105:12485-12490.
- 195. Lescure A, Deniziak M, Rederstorff M, Krol A. Molecular basis for the role of selenium in muscle development and function. Chem Biodivers 2008;5:408-413.
- 196. Rederstorff M, Allamand V, Guicheney P, et al. Ex vivo correction of selenoprotein N deficiency in rigid spine muscular dystrophy caused by a mutation in the selenocysteine codon. Nucleic Acids Res 2008; 36:237-244.
- 197. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Molec Genet 2001; 10:2851-2859.
- 198. Beltran-Valero de Bernabe D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet 2004; 41:e61.
- 199. Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998; 394:388-392.
- 200. Beltran-Valero de Bernabe D, van Bokhoven H, van Beusekom E, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet 2003; 40:845-848.
- 201. Murakami T, Hayashi YK, Noguchi S, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 2006; 60:597-602.
- 202. Godfrey C, Escolar D, Brockington M, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol 2006; 60:603-610.
- 203. Clement EM, Godfrey C, Tan J, et al. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch Neurol 2008; 65:137-141.
- 204. Biancheri R, Bertini E, Falace A, et al. POMGnT1 mutations in congenital muscular dystrophy: genotype-phenotype correlation and expanded clinical spectrum. Arch Neurol 2006;63:1491-1495.
- 205. Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 2002; 71:1033-1043.
- 206. van Reeuwijk J, Maugenre S, van den Elzen C, et al. The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat 2006; 27:453-459.
- 207. Biancheri R, Falace A, Tessa A, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 2007; 363:1033-1037.
- 208. Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Molec Genet 2003; 12:2853-2861.
- 209. van Reeuwijk J, Grewal PK, Salih MA, et al. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet 2007; 121:685-690.
Publication Dates
-
Publication in this collection
08 June 2009 -
Date of issue
June 2009
History
-
Accepted
14 Mar 2009 -
Received
31 Oct 2008