Resumo
Oxidative stress is the result of an imbalance between oxidant and antioxidant species, with predominance of oxidative species with harmful action of reactive oxygen species (ROS) and reactive nitrogen species (RNS) on cells. Changes in the levels of nitric oxide (NO•) can be the cause and/ or a result of various pathophysiological processes. The main objective of this review is to address the relationship between oxidative stress and atherosclerosis in order to better understand the main features of this disease.
oxidative stress; nitric oxide; atherosclerosis
oxidative stress; nitric oxide; atherosclerosis
REVISÃO
Relações patofisiológicas entre estresse oxidativo e arteriosclerose
Pathophysiologic relationships between oxidative stress and atherosclerosis
Danielle da Costa SilvaI; Giselle CerchiaroI, * * e-mail: giselle.cerchiaro@ufabc.edu.br ; Káthia M. HonórioII
ICentro de Ciências Naturais e Humanas, Universidade Federal do ABC, Av. dos Estados, 5001, Bl. B, 09210-170 Santo André - SP, Brasil
IICentro de Ciências Naturais e Humanas, Universidade Federal do ABC, Av. dos Estados, 5001, 09210-170 Santo André - SP / Escola de Artes, Ciências e Humanidades, Universidade de São Paulo, Av. Arlindo Bettio, 1000, 03828-000 São Paulo - SP, Brasil
ABSTRACT
Oxidative stress is the result of an imbalance between oxidant and antioxidant species, with predominance of oxidative species with harmful action of reactive oxygen species (ROS) and reactive nitrogen species (RNS) on cells. Changes in the levels of nitric oxide (NO) can be the cause and/ or a result of various pathophysiological processes. The main objective of this review is to address the relationship between oxidative stress and atherosclerosis in order to better understand the main features of this disease.
Keywords: oxidative stress; nitric oxide; atherosclerosis.
INTRODUÇÃO
A arteriosclerose é uma doença inflamatória que se destaca como principal causa de morbi-mortalidade no mundo. No Brasil, ocorrem aproximadamente 300 mil óbitos/ano, causados por doenças cardiovasculares associadas à arteriosclerose e dados ainda mais assustadores são encontrados nos Estados Unidos, onde as doenças cardiovasculares são responsáveis por cerca de 80% das mortes de pessoas com mais de 65 anos.1-3 Atualmente, as doenças inflamatórias têm sido relacionadas a um desbalanço redox e efeitos do excesso de espécies oxidantes nos organismos. Por isso, tem-se observado um enorme interesse no estudo de antioxidantes devido, principalmente, às descobertas sobre o efeito dos radicais livres no organismo.4 A oxidação é uma parte fundamental da via aeróbica e do metabolismo e, assim, os radicais livres são produzidos naturalmente ou por alguma disfunção biológica. Esses radicais livres, cujo elétron desemparelhado se encontra centrado nos átomos de oxigênio ou nitrogênio, são denominados espécies reativas de oxigênio (ROS), ou espécies reativas de nitrogênio (RNS) ambas radicalares (com um ou mais elétrons desemparelhados) ou não.5,6
No organismo, os radicais livres encontram-se envolvidos na fagocitose, regulação do crescimento celular, sinalização intercelular e síntese de substâncias biológicas importantes. No entanto, seu excesso acarreta efeitos prejudiciais, tais como a lipoperoxidação de membranas e oxidação de proteínas.4,7 ROS e RNS incluem os radicais livres, como o ânion superóxido, radicais hidroxila e peroxila, bem como espécies não radicalares, como peróxido de hidrogênio, peroxinitrito e oxigênio singlete. As ROS são produzidas continuamente na cadeia respiratória das mitocôndrias com a redução de um elétron do oxigênio e molecular. NAD(P)H oxidase, xantina oxidase, mieloperoxidase, cicloxigenase e lipoxigenase são grandes fontes enzimáticas de ROS em células de mamíferos, enquanto a radiação UV representa um exemplo de um fator ambiental de geração de ROS.8
Em altas concentrações, ROS e RNS podem danificar macromoléculas celulares, incluindo DNA, proteínas e lipídios, podendo levar à morte celular subsequente. Assim, as células possuem mecanismos de defesa e sistemas antioxidantes para detoxificação de ROS e RNS, bem como a reparação de modificações oxidativas deletérias sobre as estruturas celulares. O estresse oxidativo é, portanto, o resultado do desequilíbrio entre moléculas antioxidantes e oxidantes, causada por distúrbios na produção, distribuição ou por uma superabundância de ROS ou RNS a partir de fontes endógenas ou fatores ambientais, predominando a ação danosa das ROS e RNS sobre as células. Por causa disso, o estresse oxidativo tem sido relacionado na patogênese de diversas doenças, que variam de cardiovasculares, neurodegenerativas, alguns tipos de câncer, bem como no processo de envelhecimento.9 Vale ressaltar que as espécies reativas em geral podem ser ao mesmo tempo causa e consequência de patologias humanas associadas ao estresse oxidativo.10
Neste ponto, a arteriosclerose vem sendo estudada sob os mais variados enfoques. No entanto, estudos relacionados ao metabolismo das lipoproteínas de baixa densidade (LDL) vêm ganhando grande destaque no entendimento desta doença.11 Qual será a relação existente? Será que o estresse oxidativo é causa ou consequência do desenvolvimento da arteriosclerose? Como ocorre esta interação? Estes são alguns tópicos abordados nesta revisão.
As células danificadas por ROS e/ou RNS podem apresentar, por exemplo, modificações em sua função devido ao acúmulo de LDL oxidadas (LDLox). Esta alteração pode ser observada nas células endoteliais dos vasos sanguíneos e, também, em células fagocitárias da região endotelial afetada, sendo responsável pelo desenvolvimento da placa de ateroma formada.1
De acordo com a falta de relação explícita entre radicais livres e as doenças degenerativas, o principal objetivo desta revisão foi abordar o que tem sido feito em relação à influência do estresse oxidativo e a doença arteriosclerose, devido ao grande número de estudos que vêm sendo desenvolvidos para um melhor entendimento desta relevante doença.
ÓXIDO NÍTRICO (NO )
O óxido nítrico (NO) é um gás incolor à temperatura ambiente, habitualmente encontrado no ar atmosférico, pouco solúvel em água, sendo mais solúvel em solventes orgânicos como o n-hexano, onde apresenta uma concentração na saturação de 0,13 M.12 Dessa forma, esta molécula tende a dissolver-se seletivamente na membrana e em regiões que contêm lipídeos presentes nas células. Isto acontece, pois, a energia livre de hidratação desfavorável de uma substância apolar gerada pela ordenação das moléculas circundantes de água resulta do fato de a substância apolar ser excluída da fase aquosa. A molécula neutra de NO apresenta 11 elétrons na camada de valência e é paramagnética, devido à seguinte configuração eletrônica: (σ2s)2 (σ2s*)2 (σ2pz)2 (π2p)4 (π2p*)1.13-15 O óxido nítrico apresenta um elétron desemparelhado, sendo uma espécie radicalar (NO), o que lhe confere uma alta reatividade com meia-vida de 5 a 10 s in vitro, sendo capaz de reagir rapidamente com outros radicais importantes biologicamente, dentre eles o oxigênio molecular (O2) e o radical superóxido (O2-), podendo complexar-se também com metais de transição como o ferro, por exemplo.11-13 O significado da oxidação do NO pelo O2- é foco de diversos trabalhos na literatura.16
O NO reage com o O2 para formar dióxido de nitrogênio, NO2, tanto em fase gasosa quanto em fase aquosa. O NO2 reage com outra molécula de NO e produz trióxido de dinitrogênio, N2O3 (Equação 1), ou com o próprio NO2 (por dimerização), gerando tetróxido de dinitrogênio, N2O4 (Equação 2). Todas essas espécies reagem muito rápido com água, formando íons nitrato e nitrito.15

O N2O3 é considerado um potencial doador de nitrosônio (NO+). Este é denominado um intermediário-chave, possuindo um potencial redox de 1,60 V e podendo ser transferido para uma grande diversidade de nucleófilos presentes em biomoléculas, como as aminas, por exemplo, levando à formação de nitrito e N-nitrosaminas, respectivamente.17 O óxido nítrico também pode reagir com o ânion superóxido (O2-) e formar o ânion peroxinitrito (ONOO-) que, em pH neutro, é protonado rapidamente formando o ácido peroxinitroso (HONO2, a 25 ºC possui pKa = 6,8). Este se decompõe rapidamente, devido a sua alta instabilidade, por duas vias diferentes, produzindo dióxido de nitrogênio e radical hidroxila.15 A formação de peroxinitrito, ácido peroxinitroso e seus produtos de decomposição (OH e NO2) pode ser prejudicial às macromoléculas, pois essas espécies são capazes de oxidar lipídeos, tióis e DNA.18,19 Vale ressaltar que as metaloproteínas (proteínas que contenham um ou mais íons metálicos em sua estrutura) podem reagir com o NO, formando complexos entre o NO e o metal de transição; neste caso, pode-se citar como exemplo a hemoglobina, uma metaloproteína que possui ferro no grupo heme.20,21 Na presença do par bicarbonato/dióxido de carbono, o peroxinitrito se decompõe em parte a radical carbonato (E0 = 1,8 V, pH 7,0), importante espécie na oxidação de biomoléculas.16 A maior parte do NO gerado no sistema vascular é eventualmente removido pela interação com a hemoglobina. Como resultado, a meia-vida do complexo NO-Hb formado é menor do que 1 μs. A taxa de entrada limite do NO em eritrócitos e sua reação com a oxi-hemoglobina é estimada por meio da meia-vida do NO em sangue total, que é de cerca de 18 ms.21
SÍNTESE E AÇÃO BIOLÓGICA DO NO
O NO depende apenas de sua capacidade redox para se ligar a uma enzima ou receptor biológico. Além disso, essa espécie ainda está envolvida em uma grande variedade de vias biológicas que relatam sua importância biológica, bem como sua atuação como mensageiro, inibindo ou ativando diversas moléculas-alvo envolvidas em vários processos, além de ser sintetizado de acordo com a necessidade do organismo, espalhando-se instantaneamente até seu sítio de ação.15
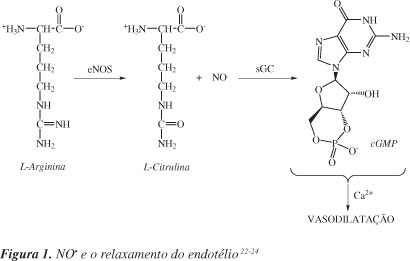
Existe uma variedade de células que utilizam a arginina para sintetizar o óxido nítrico. Nas células presentes no endotélio vascular, na presença de oxigênio molecular, a L-arginina produz o NO e L-citrulina em um processo catalisado pela enzima óxido nítrico sintase, NOS. O óxido nítrico atravessa o espaço presente entre o endotélio e o músculo liso vascular e estimula diretamente a enzima guanilato ciclase solúvel (sGC) e, consequentemente, ocorre a formação de monofosfato cíclico de guanosina (cGMP) intracelular, diminuindo os níveis de íons Ca2+ intracelular, resultando no relaxamento do endotélio, como ilustrado na Figura 1.22-24 Caso inibidores da degradação do cGMP estejam presentes (por exemplo, o sidenafil), este mensageiro intracelular continua presente na célula causando o relaxamento do músculo liso, por estimular a fosforilação de proteínas pela ação da proteína-cinase dependente de cGMP.
Quando a hipótese de que o fator de relaxamento derivado do endotélio seria o NO foi levantada por Furchgott e colaboradores em torno da segunda metade da década de 70, não se aceitava a possibilidade de uma molécula tão reativa como o NO ser responsável pela ativação da enzima sGC e ainda atuar como mediador gasoso responsável por uma variedade de fenômenos biológicos.25,26 Na década de 80, Ignarro e Furchgott comprovaram que o fator de relaxamento derivado do endotélio (EDRF) era o óxido nítrico, NO.26,27
O relaxamento dos vasos sanguíneos (vasodilatação) se mantém enquanto a propagação do NO para a musculatura lisa vascular (endotélio) estiver ocorrendo. Um aumento no fluxo de NO para o endotélio provoca maior relaxamento celular e, consequentemente, maior vasodilatação. No entanto, se a formação de NO diminui, ocorre o que se denomina de vasoconstrição moderada.23 Uma alteração nos níveis de concentração do NO, seja deficiência ou excesso, pode ser a causa de vários processos patofisiológicos.28 Em especial, a diminuição da produção de NO pode ser induzida por alguns fatores, dentre os quais estão hipertensão, diabetes e hipercolesterolemia.
A óxido nítrico sintase (NOS) é a enzima responsável pela síntese endógena do NO. Existem três tipos de NOS: uma NOS induzida (iNOS) e duas NOS constitutivas (cNOS). Elas estão presentes no citosol e são inibidas por análogos da L-arginina e requerem NADPH, flavina adenina dinucleotídeo (FAD), flavina mononucleotídeo (FMN) e heme como cofatores.23,29-31 A óxido nítrico sintase neuronal (nNOS) é uma enzima constitutiva, presente em neurônios, células epiteliais, SNC e SNP, músculo esquelético, órgão sexual masculino e outros. Atua na regulação central da pressão sanguínea, no relaxamento do músculo liso e na vasodilatação através dos nervos periféricos; também é responsável pelo fluxo sanguíneo cerebral local, está envolvida na formação da memória e participa da resposta imune.23,32,33 A iNOS é uma enzima induzida em macrófagos e contribui para a resposta celular, produção de citocinas e lipopolissacarídeos presentes no endotélio e na musculatura lisa vascular. Produz grande quantidade de NO que tem efeito citostático por inibição de enzimas contendo ferro, e causa a fragmentação do DNA; também atua em células tumorais.23,32,33 Já a eNOS é uma enzima constitutiva e produz NO no endotélio vascular sob condições basais (Figura 1); esta isoforma também pode ser encontrada em linfócitos T, plaquetas e eritrócitos. O NO liberado no lúmen vascular é um potente inibidor da adesão de leucócitos ao endotélio vascular, sendo também responsável pela pressão da regulação sanguínea e contratilidade da musculatura cardíaca.23,34
ESTRESSE OXIDATIVO E DISFUNÇÃO ENDOTELIAL
O acúmulo de ROS e RNS no endotélio pode acontecer por vias enzimáticas - (NAD(P)Hox), XO além da eNOS - e por meio de forças mecânicas (forças hemodinâmicas/ cisalhamento), chamadas de shear stress, que são produzidas a partir de um forte fluxo sanguíneo em consequência da maior pressão no endotélio vascular.21
No estresse oxidativo vascular tem-se uma superprodução de O2-, que diminui a atividade do NO reagindo com ele para formar peroxinitrito, um intermediário altamente reativo e prejudicial às biomoléculas; esta espécie forma posteriormente o radical hidroxila e a reação ocorre independente da presença do metal de transição (Esquema 2).8,35-40
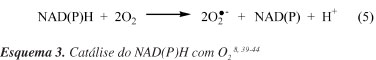
Considera-se o sistema NADH/NAD(P)Hox a maior fonte de O2- nas membranas das células endoteliais.41-44 As NAD(P)Hox são enzimas que catalisam a redução de O2 usando NAD(P)H como doador de elétron (Esquema 3). Quando estas enzimas são ativadas por forças hemodinâmicas e por agonistas vasoativos, em especial pela angiotensina II (Ang II ou AII), as NAD(P)Hox vasculares (isoformas encontradas na célula endotelial) produzem O2- extra e intracelularmente, enquanto que a NAD(P)Hox produz O2- apenas extracelularmente.8,39-44
A xantina oxidase (XO) está presente no endotélio, sendo uma molibdoenzima capaz de catalisar a oxidação de xantina e hipoxantina via O2, gerando O2-, H2O2 e urato, sob condições inflamatórias. O subproduto O2-, sob a ação da superóxido dismutase (SOD), gera H2O2 e O2 (via uma reação de dismutação de dois radicais superóxido), e o subproduto H2O2 em uma segunda reação possível para produzir H2O e O2 pode ser catalisado pela enzima glutationa peroxidase (GSH-Px) ou catalase, ou ainda a reação na presença de metal de transição, como Fe2+ ou Cu1+, gerando o radical OH.8,42,45,46
Por se tratar de uma doença cardiovascular, um dos mecanismos mais estudados na doença ateriosclerótica está relacionado com a lipoperoxidação a partir da produção da angiotensina II (AngII) do sistema renina-angiotensina-aldosterona (SRAA). A AngII ativa a produção de NAD(P)Hox e a XO diminui a síntese de NO por alterar a síntese de eNOS. 8,36,39,47 De acordo com a hipótese de modificação oxidativa da aterogênese, metabólitos do oxigênio contribuem para a progressão do processo aterosclerótico, modificando oxidativamente a lipoproteína de baixa densidade (LDL). A ativação das células inflamatórias resulta no aumento dos leucócitos no sangue, o que favorece a aterogênese, promovendo a oxidação do LDL. Além disso, é razoável que o aumento da regulação dos sistemas antioxidantes desencadeie um contínuo processo inflamatório, o que conduz a uma diminuição na capacidade antioxidante.48
Um dado relevante é que todos os tipos de células presentes nas paredes dos vasos (células endoteliais, células musculares lisas e fibroblastos) expressam a enzima NOS e podem liberar O2- e H2O2. Primeiro, considerando as células endoteliais, diversas fontes contribuem para a sua produção de ROS extracelular: aminas oxidases produzem H2O2; xantina oxidase pode se ligar a células endoteliais superficiais, gerando O2- e H2O2; NOS endotelial produz O2-, em determinadas circunstâncias. O significado fisiológico da produção de O2- endoteliais extracelular é incerto, e é provável que O2- e NO antagonizem umas das outras ações como parte de um mecanismo normal vasorregulatório, uma visão suportada pelo fato de que NOS em camundongos diminuiu a pressão arterial. Evidentemente, o ONOO- resultante teria de ser eliminado de forma segura. Células do músculo liso vascular e fibroblastos também contêm NOS.21
Fibroblastos e células do músculo liso podem responder à ROS pela proliferação, provocando uma hipertrofia da parede do vaso. Citocinas podem acelerar a proliferação de fibroblastos e trombina, além de estimular a proliferação das células musculares lisas; ROS estão envolvidas em ambos os processos. O conceito de que o ânion O2- desempenha um papel na vasorregulação normal é fortalecido pela observação de que a angiotensina II estimula a produção de O2- por todos os três tipos de células, em parte pelo aumento da fosforilação e por promover um conjunto de oxidações. Estudos de administração da angiotensina II em animais demonstraram aumento nos níveis de F2-isoprostanos, indicativo de aumento do estresse oxidativo.21 A produção de O2- vascular é aumentada em hipercolesterolemia e contribui para a arteriosclerose. Estudos em camundongos mostram que ele está envolvido na angiogênese, em locais de lesão tecidual. A hipertensão não só pode resultar do excesso de ROS, mas pode ser a causa da formação de mais O2-; estresse mecânico excessivo em vasos sanguíneos aumenta os níveis de NOS, levando a uma diminuição nos níveis de tetra-hidrobiopterina, um processo em que a NOS pode contribuir para a formação de mais ROS. Quando a tetra-hidrobiopterina se encontra em baixa quantidade, ela facilita a produção de O2- por eNOS.21
A produção da AngII ativa as enzimas XO e NAD(P)Hox, causando modificações na atividade da enzima óxido nítrico sintase endotelial (eNOS). O desequilíbrio entre a formação de NO e O2- reforça a oxidação do LDL (LDLox), acarretando o acúmulo de lipídeos nas paredes das artérias (Figura 2), dando início à formação de ateromas.8,47 Aliados a este fator, uma menor produção de NO e maior produção de ONOO- geram o comprometimento do relaxamento do endotélio, causando a denominada disfunção endotelial, o que favorece um aumento na atividade da Endotelina-1 (ET-1), causando a vasoconstrição e o desenvolvimento progressivo das células endoteliais.5,40,47 O revestimento endotelial do sistema cardiovascular é altamente sensível a cisalhamento hemodinâmico, que atua na parede superficial do lúmen vascular, na direção do fluxo sanguíneo. Este processo de cisalhamento do endotélio é sistematicamente comprometido por hiperlipidemia, hipertensão, diabetes e doenças inflamatórias. A evolução dos fluxos relacionados com as respostas no endotélio é uma das condições essenciais para o funcionamento eficiente do sistema vascular.49 Forças hemodinâmicas, em especial o cisalhamento, são reguladoras de muitos eventos fisiológicos e aspectos patológicos da função endotelial no sistema cardiovascular e as respostas endoteliais são sensíveis a variações nas características que geram fluxo de cisalhamento, regiões com oscilação de cisalhamento e reversão de fluxo correspondem a alterações patológicas na parede da artéria e são um fator de susceptibilidade para a arteriosclerose.49
Quando o cisalhamento deforma o endotélio, uma perturbação mecânica é comunicada através do citoesqueleto para várias áreas de transdução mecânica, que incluem a célula-matriz, sítios de adesão e membrana nuclear. Como efeitos benéficos sistêmicos têm-se a manutenção hemodinâmica arterial dentro de limites normais por meio de terapias anti-hipertensivas, exercícios regulares para promover a adaptação contínua, remodelação e inibição de células causadoras da disfunção endotelial.49
CARACTERIZANDO A ARTERIOSCLEROSE
A arteriosclerose é caracterizada pela inflamação crônica na parede arterial.50 É uma doença progressiva que pode ser causada por diversos fatores, podendo iniciar-se ainda na infância ou na adolescência. Pode-se dizer que esta patologia é o resultado de uma série de respostas celulares altamente específicas. A grande quantidade de células inflamatórias, lipídeos e elementos fibrosos que se depositam na parede das artérias são os responsáveis pela formação de placas ou estrias gordurosas e que geralmente ocasionam a obstrução das mesmas, denominadas de ateromas. A formação de trombos é frequentemente desencadeada por placas que se rompem. As lipoproteínas, em especial as lipoproteínas de baixa densidade (LDL), vêm ganhando um grande destaque na origem da doença aterosclerótica. Tal situação pode resultar da interação de várias forças, incluindo irregularidades nutricionais e metabólicas, dentre as quais podem ser citadas hipercolesterolemia, hipertensão arterial e diabetes mellitus.51-55
A resposta inflamatória no início da doença aterosclerótica é precedida por alterações funcionais nas células endoteliais, linfócitos T, macrófagos e células do músculo liso. Estas células, quando ativadas, proporcionam, principalmente, um acúmulo de lipídeos. Além disso, a resposta inflamatória também pode ser induzida pelo estresse oxidativo, especialmente pela oxidação do LDL, processo denominado de peroxidação lipídica ou lipoperoxidação.51,52,54,56,57 A formação de ateromas resulta, consequentemente, em uma lesão endotelial. Esta lesão permite a entrada de mais LDL na parede do vaso sanguíneo seguido pelo desenvolvimento de macrófagos na parede do vaso. Os macrófagos ali presentes secretam citocinas que estimulam o aumento da resposta inflamatória local, havendo aumento de citocinas e quimiocinas, ambas responsáveis pelo recrutamento dos leucócitos. Linfócitos também podem entrar na parede do vaso, sendo atraídos pelas citocinas, estimulando a formação de anticorpos,21 isto pode manter e modificar as lipoproteínas localizadas no endotélio vascular, aumentando a sua fagocitose em macrófagos através das classes A e B receptores, no caso, de lipoproteína oxidada de baixa densidade (LDLox), causando a produção de citocinas como o fator de necrose tumoral (TNF) - alfa, interleucina (IL) -1 beta, IL-4, IL-6, IL-12 e IL-18, entre outros fatores. Com a secreção de citocinas ocorre a ativação das células T, gerando linfócitos T (Th1), capazes de reconhecer a LDLox e proteínas de choque térmico como antígenos, amplificando a resposta inflamatória.58 As metaloproteínas presentes (hemoglobina, mioglobina, por exemplo), podem danificar a "cápsula" fibrosa formada e enfraquecer a placa de ateroma. Como os macrófagos não podem limitar a absorção de colesterol, eles dependem, portanto, de vias de efluxo de colesterol para evitar a acumulação desta substância que produz a formação de estrias gordurosas, sendo o primeiro passo para a arteriosclerose.59
Os macrófagos estão ocupando de certa forma um papel central na doença, pois foi descoberto recentemente que são importantes reguladores do processo inflamatório da arteriosclerose. A diferenciação dos macrófagos e sua função podem ser moduladas por uma classe de fatores de transcrição chamados receptores nucleares, dentre os quais se pode citar PPARs e LXRs.60 Os PPARs e LXRs são conhecidos como fatores de transcrição ligante-regulados que desempenham funções de regulação central na absorção de lipídeos, metabolismo e de efluxo de colesterol. Eles também são importantes por estarem envolvidos na regulação das reações inflamatórias. PPARs e LXRs são ativados por ácidos graxos, LDLox, metabólitos do ácido araquidônico, como eicosanoides, prostaglandinas, leucotrienos e colesterol oxidado, respectivamente. A maioria destes ligantes naturais é derivada do metabolismo dos ácidos graxos insaturados e do colesterol. Desta forma, alterações no consumo de lipídeos, bem como o seu processamento, podem influenciar a atividade transcricional dos genes regulados pelos receptores PPAR e LXR.60
LIPOPEROXIDAÇÃO
Os componentes celulares estão sujeitos à ação das RNS e ROS. No entanto, a membrana celular é uma das mais atacadas por espécies reativas devido à peroxidação lipídica, pois ocasiona modificações tanto na permeabilidade das membranas celulares quanto em sua estrutura. Este processo também está intimamente associado aos mecanismos de câncer e de envelhecimento, por exemplo. Todavia, nem sempre o processo de lipoperoxidação será prejudicial, bem como a formação de ROS e RNS, devido à importância de seus produtos na resposta inflamatória. Por outro lado, o excesso de tais produtos também pode ser muito prejudicial às biomoléculas.61-64
Vários experimentos realizados em células cultivadas sob estresse oxidativo indicam a suscetibilidade do colesterol em sofrer peroxidação lipídica via celular. Considera-se também que ocorre uma alteração nos níveis plasmáticos dos produtos de oxidação do colesterol, por meio do aumento não só da peroxidação lipídica no plasma, mas também da peroxidação lipídica celular.65 As moléculas de LDL são facilmente oxidadas à LDLox quando o estresse oxidativo está presente. Estas moléculas provocam o ataque ao tônus endotelial, provocando uma resposta inflamatória nesta região. LDLox induzem a expressão de moléculas de adesão e ligação posterior às células inflamatórias, bem como sua migração no espaço subendotelial. Os resultados disso são a disfunção endotelial, agregação plaquetária, expressão da metaloproteinase e trombogênese. Com o acúmulo progressivo de LDLox, macrófagos modulam seu fenótipo, tornando-se as chamadas células espumosas. As células espumosas são os principais componentes das estrias gordurosas, sendo este o primeiro passo na formação da placa ateromatosa, e provocam reação antigênica de linfócitos T que iniciam ou aumentam a resposta imunológica. Outra evidência apoiando a oxidação excessiva inclui o aumento dos níveis de peroxidação lipídica, como a presença do LDLox. Sabe-se também que o equilíbrio normal entre a formação de NO e ROS é interrompido na aterosclerose. NO inibe a ativação da xantina oxidase e NADPH oxidase, mantendo a homeostase O2-/NO normal. Assim, com mais NO que está sendo convertido à peroxinitrito, isso leva a uma perda normal de NO, que compromete a função endotelial, e o comprometimento da função endotelial é um indicador precoce de aterosclerose.66
A peroxidação lipídica se inicia com o sequestro do hidrogênio do ácido graxo poli-insaturado (LH) da membrana lipídica. Este sequestro pode ser realizado tanto pelo alcoxila (LO ) ou pelo radical OH, seguido da formação do L (radical lipídico) e H2O (Esquema 4), onde o radical OH é reconhecido como a espécie iniciadora e a mais importante desse processo, podendo ser gerado a partir da decomposição do ONOO-, por exemplo.61 Na etapa seguinte, a forma L reage com o O2, com a consequente formação do radical peroxila (LOO); este, por sua vez, sequestra novo hidrogênio do LH originando mais uma vez L, por isso se diz que esta é uma reação em cadeia. O processo de lipoperoxidação termina quando os radicais L e LOO, gerados nas etapas anteriores, estendem-se até se autodestruírem (Esquema 5), onde L representa o lipídeo.61
A peroxidação lipídica pode ser catalisada por íons ferro via conversão de hidroperóxidos lipídicos (LOOH) em radicais extremamente reativos (peroxila (LOO) e alcoxila (LO)), os quais dão início a uma nova cadeia de reações. Estas podem ser lentas ou rápidas, isto vai depender da valência do ferro (Esquema 6). Como exemplo pode-se citar: se o ferro for o agente oxidante e o eritrócito a célula-alvo, a parte lipídica da membrana eritrocitária também estará susceptível à agressão oxidativa, sendo que os produtos deste processo de oxidação lipídica podem levar ao estresse oxidativo intracelular.61,67
As lipoproteínas de baixa densidade oxidadas ou alteradas impedem a saída de macrófagos da lesão, além de torná-los capazes de fagocitar grandes quantidades de lipídeos. A LDLox alterada é obtida por receptores que não estão expostos a este tipo de controle.68 Por sua vez, a obtenção demasiada de LDLs alteradas (LDL oxidada) por macrófagos transforma estas células em células "espumosas" que participam na formação das placas ateroscleróticas.68
CONSIDERAÇÕES FINAIS
Embora a causa exata no que diz respeito ao desenvolvimento da arteriosclerose ainda seja desconhecida, sabe-se que esta doença envolve diversos mecanismos fisiopatológicos. A patologia descrita neste trabalho é lenta e progressiva e está intimamente relacionada a vários fatores de risco, dentre eles hipercolesterolemia, hipertensão arterial e diabetes mellitus. A exposição do organismo a estes fatores ocasiona o comprometimento da função do endotélio vascular e diminui a biossíntese de NO.69
Estudos relativos ao metabolismo das lipoproteínas de baixa densidade (LDL) são os que mais têm se desenvolvido.6 Deste modo, conclui-se que os caminhos pró-oxidativos e pro-inflamatórios no endotélio vascular têm sido implicados na iniciação e progressão da arteriosclerose. Recentes pesquisas revelaram evidências da IL-4 (interleucina-4), considerada anti-inflamatória, ser na verdade pró-inflamatória no endotélio vascular, tendo um papel importante na arteriosclerose por mediar a formação de ROS.70 No processo inflamatório, a arteriosclerose é o resultado da disfunção endotelial em função do estresse oxidativo relacionado a fatores de risco. A inflamação e o estresse oxidativo são mediadores patogênicos de várias doenças, contribuindo para o dano tecidual em várias situações, dentre as quais podemos citar lesões no cérebro, rins e coração.64,71
Por fim, vale ressaltar que as ROSs ou RNSs podem efetivamente ser causa e consequência de patologias humanas associadas ao estresse oxidativo e a utilização de fármacos que contenham antioxidantes deve ser cuidadosamente supervisionada no tratamento de doenças relacionadas ao estresse oxidativo.
ABREVIATURAS
ROS - Espécies Reativas de Oxigênio
RNS - Espécies Reativas de Nitrogênio
LDL - Lipoproteínas de baixa densidade
LDLox - Lipoproteínas de baixa densidade oxidada
sGC - Guanilato ciclase solúvel
cGMP - Monofosfato cíclico de guanosina
eNOS - Enzima óxido nítrico sintase endotelial
EDRF - Fator de relaxamento derivado do endotélio
ET-1 - Endotelina-1
GSH-Px - Enzima glutationa peroxidase
iNOS - Enzima óxido nítrico sintase induzida
cNOS - Enzima óxido nítrico sintase constitutiva
FAD - Flavina adenina dinucleotídeo
FMN - Flavina mononucleotídeo
XO - Xantina oxidase
NAD(P)Hox - NADPH oxidase
SOD - Superóxido dismutase (Cu,Zn-SOD)
Ang II ou AII - Angiotensina II
PPARs - Peroxisomal proliferator-activated receptor
LXRs - Liver X receptors
AGRADECIMENTOS
À FAPESP, CNPq, Fundação Universidade Federal do ABC e CAPES pelo auxílio financeiro.
-
1Giroldo, M. L.; Alves, A. S.; Baptista, F.; Sábios-Rev. Saúde e Biol. 2007, 2, 32.
-
2Saini, H. K.; Xu, Y. J.; Arneja, A. S.; Tappia, P. S.; Dhalla, N. S.; J. Cell. Mol. Med. 2005, 9, 818.
-
3Machado, D. F.; Ferreira, C. L. L. F.; Costa, N. M. B.; Oliveira, T. T.; Ciência Tecnol. Aliment. 2003, 23, 270.
-
4Barreiros, A. L. B. S.; David, J. M.; David, J. P.; Quim. Nova 2006, 29, 113.
-
5Visioli, F.; Keaney Jr., J. F.; Halliwell, B.; Cardiovasc. Res. 2000, 47, 409.
-
6Finkel, T.; Holbrook, N. J.; Nature 2000, 408, 239.
-
7Husain, S. R.; Cillard, J.; Cillard, P.; Phytochemistry 1987, 26, 2489.
-
8Steinbrenner, H.; Sies, H.; Biochim. Biophys. Acta 2009, 1790, 1478.
-
9Sies, H.; Am. J. Med. 1991, 91, S31.
-
10Vasconcelos, S. M. L.; Goulart, M. O. F.; Silva, M. A. M.; Gomes, A. C. M.; Rev. Bras. Hipertensão 2007, 14, 269.
-
11Ribeiro Jorge, P. A.; Arq. Bras. Cardiol. 1997, 68, 129.
-
12Shaw, A. W.; Vosper, A. J.; J. Chem. Soc., Faraday Trans. 1977, 73, 1239.
-
13Butler, A. R.; Williams, D. L. H.; Chem. Soc. Rev. 1993, 22, 233.
-
14Queiroz, S. L.; Batista, A. A.; Quim. Nova 1999, 22, 584.
-
15Barreto, R. L.; Correia, C. R. D.; Muscará, M. N.; Quim. Nova 2005, 28, 1046.
-
16Augusto, O.; Bonini, M. G.; Amanso, A. M.; Linares, E.; Santos, C. C. X.; Menezes, S. L.; Free Radical Biol. Med. 2002, 32, 841.
-
17Wink, D. A.; Darbyshire, J. F.; Nims, R.; W.; Saavedra, J. E.; Ford, P. C.; Chem. Res. Toxicol. 1993, 6, 23.
-
18Gatti, R. M.; Radi, R.; Augusto, O.; FEBS Lett. 1994, 348, 287.
-
19King, P. A.; Jamison, E.; Strahs, D.; Anderson, V. E.; Brenowitz, M.; Nucleic Acids Res. 1993, 21, 2473.
-
20McCleverty, J. A.; Chem. Rev. 1979, 79, 53.
-
21Halliwell, B.; Gutteridge, J. M. C.; Free Radicals in Biology and Medicine, 4th ed., Oxford University Press: Oxford, 2007, p. 53, 426 e 497.
-
22Palmer, R. M. J.; Ashton, D. S.; Moncada, S.; Nature 1988, 333, 664.
-
23Cerqueira, N. F.; Yoshida, W. B.; Acta Cirurg. Bras. 2002, 17, 417.
-
24Lyons, C. R.; Adv. Immunol. 1995, 60, 323.
-
25Ignarro, L. J.; Annu. Rev. Pharmacol. Toxicol. 1990, 30, 535.
-
26Furchgott, R. F.; Zawadzki, J. V.; Nature 1980, 288, 373.
-
27Ignarro, L. J.; Buga, G. M.; Wood, K. S.; Byrns, R. E.; Chaudhuri, G.; Medical Sciences 1987, 84, 9265.
-
28Migliori, L.; Coppedè, F.; Mutat. Res. 2009, 674, 73.
-
29Kiechle, F. L.; Malinski, T.; Am. J. Clin. Pathol. 1993,100, 567.
-
30Wong, G. K. T.; Marsden, P. A.; Nefron. Dial. Transplant. 1996, 11, 215.
-
31Moncada, S.; Palmer, R. M. J.; Higgs, E. A.; Pharmacol. Rev. 1991, 43, 109.
-
32Förstermann, U.; Ei, C.; Pollock, J. S.; Nakane, M.; Schwarz, Z. P.; Gath, I; Kleinert, H.; Hypertension 1994, 23, 1121.
-
33Viaro, F.; Nobre, F.; Evora, P. R. B.; Arq. Bras. Cardiol. 2000, 74, 365.
-
34Vanni, D. S.; Horstmann, B.; Benjo, A. M.; Daher, J. P. L.; Kanaan, S.; Sleiman, M.; J. Bras. Patol. Med. Lab. 2007, 43, 181.
-
35McIntyre, M.; Bohr, D. F.; Dominiczak, A. F.; Hypertension 1999, 34, 539.
-
36Ward, N. C.; Hodgson, J. M.; Puddey, I. B.; Mori, T. A.; Beilin, L. J.; Croft, K. D.; Free Rad. Biol. Med. 2004, 36, 226.
-
37Laurindo, F. R. M.; Souza, H. P.; Luz, P. L.; Rev. Soc. Cardiol. Estado de São Paulo 1998, 8, 344.
-
38Lassègue, B.; Griendling, K. K.; Am. J. Hypertension 2004, 17, 852.
-
39Paravicini, T. M.; Touyz, R. M.; Cardiovasc. Res. 2006, 71, 247.
-
40Portaluppi, F.; Boari, B.; Manfredini, R.; Curr. Pharm. Design 2004, 10, 1695.
-
41Beswick, R. A.; Dorrance, A. M.; Leite, R.; Webb, R. C.; Hypertension 2001, 38, 1107.
-
42Griendling, K. K.; Fitzgerald, G. A.; Circulation 2003, 108, 2034.
-
43Griendling, K. K.; Fitzgerald, G. A.; Circulation 2003, 108, 1912.
-
44Zalba, G.; San José, G.; Moreno, M. U.; Fortuño, M. A.; Fortuño, A.; Beaumont, F. J.; Díez, J.; Hypertension 2001, 38, 1935.
-
45Wassmann, S.; Wassmann, K.; Nickening, G.; Hypertension 2004, 44, 381.
-
46Touyz, R. M.; Curr. Hypertens. Rep. 2000, 2, 98.
-
47Sampaio, W. O.; Santos, R. A. S.; Rev. Bras. Hipertensão 2004, 11, 67.
-
48Rocha-Pereira, P.; Santos-Silva, A.; Rebelo, I.; Figueiredo, A.; Quintanilha, A.; Teixeira, F.; Clinica Chimica Acta 2001, 303, 33.
-
49Davies, P. F.; Nature Clin. Pract. 2009, 6, 16.
-
50Koenen, R. R.; Hundelshausen, P. V.; Nesmelova, I. V.; Zernecke, A.; Liehn, E. A.; Sarabi, A.; Kramp, B. K.; Piccinini, A. M.; Paludan, S. R.; Kowalska, A.; Kungl, A. J.; Hackeng, T. M.; Mayo, K. H.; Weber, C.; Nature Med. 2009, 15, 97.
-
51Gottlieb, M. G. V.; Bonardi, G.; Moriguchi, E. H.; Sciencia Medica 2005, 15, 203.
-
52Libby, P.; Nature 2002, 420, 868.
-
53Stein, O.; Thiery, J.; Stein, Y.; Atherosclerosis 2002, 160, 1.
-
54Hulthe, J.; Fagerberg, B.; Atheroscler. Thromb. Vasc. Biol. 2002, 22, 1162.
-
55Bensinger, S. J.; Tontonoz, P.; Nature 2008, 454, 470.
-
56Nicoletti, A.; Caligiuri, G.; Hansson, G. K.; J. Intern. Med. 2000, 247, 397.
-
57Kunsch, C.; Medford, R. M.; Circ. Res. 1999, 85, 753.
-
58Rodríguez, G.; Mago, N.; Rosa, F.; Investigation Clinica 2009, 50, 109.
-
59Pennings, M.; Meurs, I.; Ye, D.; Out, R.; Hoekstra, M.; Berkel, T. J. C. V.; Eck, M. V.; FEBS Lett. 2006, 580, 5588.
-
60Szanto, A.; Rõszer, T.; FEBS Lett. 2008, 582, 106.
-
61Ferreira, A. L. A.; Matsubara, L. S.; Rev. Ass. Med. Bras. 1997, 43, 61.
-
62Mello Filho, A. C.; Hoffman, M. E.; Meneghini, R.; Biochem. J. 1983, 218, 273.
-
63Shan, X.; Aw, T. Y.; Jones, D. P.; Pharmacol. Ther. 1990, 47, 61.
-
64Halliwell, B.; Gutteridge, J. M.C.; Enzymology 1990, 186, 1.
-
65Saito, Y.; Yoshida, Y.; Niki, E.; FEBS Lett. 2007, 581, 4349.
-
66Kao, M. P. C.; Ang, D. S. C.; Pall, A.; Struthers, A. D.; J. Hum. Hypert. 2010, 24, 1.
-
67Rice-Evans, C.; Baysal, E.; Flynn, D.; Kontoghiorghes, G.; Biochem. Soc. Trans. 1986, 14, 368.
-
68De França, T. A.; De França, N. A. A. C.; Pediatria 2001, 1, 10.
-
69Napoli, C.; Ignarro, L. J.; Nitric Oxide Biol. Chem. 2001, 5, 88.
-
70Lee, Y. W.; Kim, P. H.; Lee, W. H.; Hirani, A. A.; Biomol. Therap 2010, 18, 135.
-
71Satoh, K.; Nigro, P.; Matobal, T.; O'Dell, M. R.; Cuil, Z.; Shil, X.; Mohan, A.; Yan, C.; Abe, J.-I.; Illig, K. A.; Berk, B. C.; Nature Med. 2009, 15, 649.
Datas de Publicação
-
Publicação nesta coleção
02 Mar 2011 -
Data do Fascículo
2011