Resumo
Presently glycerol is considered a co-product of biodiesel industry. As the biodiesel production is exponentially increasing, glycerol generated from the transesterification of vegetable oils and fats is also being produced on a large scale, and turned out to be essential seeking for novel alternatives to the consumption of the extra volume, in crude and/or as derivatives high added value. This review mainly deals with chemical and enzymatic transformations of glycerol to obtain chiral building blocks for synthesis of pharmaceuticals and natural products.
glycerol; biodiesel; stereoselective synthesis
glycerol; biodiesel; stereoselective synthesis
REVISÃO
Glicerol: um breve histórico e aplicação em sínteses estereosseletivas
Glycerol: a brief history and their application in stereoselective syntheses
Adilson Beatriz* * e-mail: adilson.beatriz@ufms.br ; Yara J. K. Araújo; Dênis Pires de Lima
Departamento de Química, Centro de Ciências Exatas e Tecnologia, Universidade Federal de Mato Grosso do Sul, Av. Senador Filinto Müller, 1555, 79074-460 Campo Grande - MS, Brasil
ABSTRACT
Presently glycerol is considered a co-product of biodiesel industry. As the biodiesel production is exponentially increasing, glycerol generated from the transesterification of vegetable oils and fats is also being produced on a large scale, and turned out to be essential seeking for novel alternatives to the consumption of the extra volume, in crude and/or as derivatives high added value. This review mainly deals with chemical and enzymatic transformations of glycerol to obtain chiral building blocks for synthesis of pharmaceuticals and natural products.
Keywords: glycerol; biodiesel; stereoselective synthesis.
INTRODUÇÃO
O uso de matérias-primas de fontes renováveis pela indústria é essencial para o desenvolvimento sustentável da sociedade moderna.1 Por motivos óbvios, a necessidade de se utilizar de processos catalíticos "verdes" para converter fontes biorrenováveis em commodities químicos, dentre eles os biocombustíveis ecologicamente corretos, está mobilizando cientistas das indústrias e universidades de todo o mundo. Entre os combustíveis renováveis mais promissores destaca-se o biodiesel. Este produto é um combustível alternativo limpo, produzido pela reação de um óleo ou gordura com um álcool que reduz a viscosidade.1,2 Nessa reação, chamada de transesterificação, são formados ésteres metílicos ou etílicos de ácidos graxos (biodiesel) e glicerol. Teoricamente, para cada 3 mols de ésteres metílicos (ou etílico) é gerado 1 mol de glicerol; aproximadamente 10% da massa total do produto.1 No Brasil, existem dezenas de espécies vegetais que podem ser utilizadas para fabricação do biodiesel, tais como mamona, dendê (palma), girassol, babaçu, amendoim, pinhão manso e soja, dentre outras,3 além de sebo bovino.4
Em 2004 foi lançado, no Brasil, pelo Governo Federal, o Programa Nacional de Produção e Uso de Biodiesel (PNPB), que objetiva incentivar a produção e uso do mesmo, com enfoque na obtenção de combustíveis alternativos aos combustíveis fósseis e no desenvolvimento econômico do país (Portal do Biodiesel).5 Em 13/1/2005 foi publicada a Lei 11.097, que dispõe sobre a introdução do biodiesel na matriz energética brasileira. A lei tornou obrigatória a adição de 2% de biodiesel ao diesel (B2) até 2008 e a adição de 5% (B5) até 2013.5,6 Já em 2008 o diesel comercializado no Brasil passou a conter 3% de biodiesel (B3) e, segundo Mota et al.,7,8 as projeções mostram que o excedente de glicerol seria da ordem de 100 mil t/ano no mercado brasileiro, com a entrada do B3 nesse ano, muito além da produção tradicional, na faixa de 30 mil t/ano. As previsões para 2013, com a introdução do B5 são de uma produção de 250 mil t/ano de glicerol.8
De acordo com o programa da União Europeia, até o final de 2010 os combustíveis da frota de veículos dessa região deverão conter, no mínimo, 5,75% de biocomponentes de fontes renováveis. Se esse objetivo se cumprir, a demanda europeia por biodiesel poderá aumentar para mais de 10 milhões de t/ano até 2010, gerando cerca de 1 milhão de t/ano de glicerol como coproduto. Além disso, se os Estados Unidos substituírem 2% do diesel convencional por biodiesel (B2), em 2012 como previsto, quase 365 mil toneladas extras de glicerol seriam acrescentadas no mercado.1 Dados recentes liberados pelo Departamento de Energia dos Estados Unidos (DOE), sobre a disponibilidade mundial atual de matéria-prima oriunda de soja, milho, caixa de gordura de esgoto e sebo, mostram que 5,8 bilhões de L de biodiesel podem ser produzidos em todo o mundo.1,9 Projeções sobre a produção mundial de glicerol até 2010 apontam para 1,2 milhões de t/ano, devido ao aumento da produção de biodiesel.1
Portanto, é consenso na comunidade científica e nas indústrias do setor que a colocação do glicerol é um sério problema para a produção de biodiesel em grandes quantidades e é fundamental buscar alternativas para o consumo desse volume extra de glicerol, na forma bruta e/ou como derivados de alto valor agregado, viabilizando economicamente o aumento da produção de biodiesel.7,8
PROPRIEDADES DO GLICEROL
Quimicamente o glicerol (1) é um tri-álcool com 3 carbonos, tendo como nome sistemático (IUPAC) 1,2,3-propanotriol (Figura 1), é um líquido incolor, com gosto adocicado, sem cheiro e muito viscoso, derivado de fontes naturais ou petroquímica. O nome glicerol deriva da palavra grega glykys, doce.
Glicerol é uma das mais versáteis e valiosas substâncias químicas conhecidas para o homem.10 Comercialmente, o glicerol recebe, frequentemente, o nome de glicerina. Foi preparada pela primeira vez por Carl W. Scheele (químico sueco), em 1779, mediante o aquecimento do óleo de oliva com litargírio (PbO, usado no esmalte para cerâmicas). Na lavagem com água, obtém-se uma solução adocicada, que dá, com a evaporação da água, um líquido pesado e viscoso; seu descobridor denominou-o "o princípio doce das gorduras". Em 1846, o químico italiano Ascanio Sobrero produziu pela primeira vez a nitroglicerina e, em 1867, Alfred Nobel absorveu-a em diatomita, tornando seguro o seu manuseio como dinamite.11 Estas descobertas provocaram aumento na demanda da glicerina. A demanda era, em parte, satisfeita pelo desenvolvimento de um método, em 1870, de recuperação da glicerina e do sal das lixívias do sabão. Desde 1948, aproximadamente, o glicerol é produzido sinteticamente a partir de matérias-primas petroquímicas. A Tabela 1 mostra algumas propriedades físico-químicas12 do glicerol puro.
A presença de três grupos hidroxila na estrutura do glicerol é responsável pela solubilidade em água e sua natureza higroscópica. É uma molécula altamente flexível formando ligações de hidrogênio tanto intra como intermoleculares. De acordo com estudos teóricos, usando métodos de teoria de densidade funcional (DFT), existem 126 possíveis confôrmeros para o glicerol.13 Os métodos usados nesses estudos indicam que as contribuições entálpicas e entrópicas para a energia livre de Gibbs são importantes para uma determinação precisa das preferências conformacionais e energéticas do glicerol. No confôrmero de menor energia, denominado de gG'g,g'Gg (γγ), os grupos hidroxila formam uma estrutura cíclica com três ligações de hidrogênio internas, com os comprimentos de ligação indicados na Figura 2(a). Esta geometria fornece o ponto de partida para mecanismos de várias reações químicas com aplicações práticas. Na fase aquosa, a molécula de glicerol é estabilizada por uma combinação de ligações de hidrogênios intramoleculares e solvatação intermolecular das hidroxilas. Levando em conta a solvatação, o confôrmero com duas ligações de hidrogênio intramolecular, mostrado à direita da Figura 2, é o mais estável energeticamente.13
PRODUÇÃO DE GLICEROL
Desde o final dos anos 40 o glicerol tem sido produzido a partir da epicloridrina (Esquema 1) obtida a partir do propileno (e este de combustíveis fósseis). Hoje, no entanto, plantas de produção de glicerol estão fechando e plantas que usam glicerol como matéria-prima estão sendo abertas (inclusive para produção da epicloridrina), como resultado do grande excedente de glicerol obtido como coproduto da produção de biodiesel.14 Uma quantidade significativa de glicerol também pode ser produzida a partir do álcool alílico,15 por via fermentativa,15,16 e através de hidrogenação de carboidratos.17
Como antecipado, o biodiesel é produzido por uma reação de transesterificação entre óleo vegetal e metanol, catalisada por KOH ou NaOH. É uma reação de equilíbrio dinâmico, onde 1 mol de triglicerídeo reage com 3 mols de metanol (ou etanol). Para se obter rendimento satisfatório dos produtos, é adicionado grande excesso de álcool em relação ao triglicerídeo, deslocando o equilíbrio para a direita da equação, de acordo com o Princípio de Le Chatelier. Melhores resultados são obtidos quando se usam relações em mol entre 9:1 e 12:1.4
A separação da glicerina do biodiesel metílico ocorre por decantação, facilitando o processo de purificação. Na transesterificação, como o metanol é adicionado em excesso, forma-se duas fases: uma rica em metanol e uma rica em biodiesel. A glicerina distribui-se entre essas duas fases, predominando, porém, na fase metanólica. No entanto, quando se utiliza etanol, a separação das fases dos produtos finais é complicada.
A glicerina resultante da fabricação do biodiesel (glicerina bruta vegetal) apresenta cerca de 20% de impurezas.8 Dependendo do grau de pureza, a diferença no preço do produto é significativa. A cotação da glicerina bruta, em 2008, estava em torno de R$ 105,00/t, a bidestilada (96%) era de R$ 2.100,00/t, enquanto que a glicerina farmacêutica (>99,5%) era comercializada por R$ 2.500,00/t.18 As principais impurezas presentes na glicerina do biodiesel são catalisador, álcool, ácidos graxos, sais e água. Estas impurezas dependem do tipo da oleaginosa e do tipo de catálise empregada na produção do biodiesel. Consequentemente, a glicerina bruta tem poucas aplicações diretas. A purificação da glicerina bruta pode ser feita por destilação sob pressão reduzida, resultando num produto transparente. Alternativamente, uma solução econômica para a purificação do coproduto do biodiesel combina eletrodiálise (membranas) e nanofiltros, fornecendo um líquido transparente com baixo teor de sais. Esta glicerina parcialmente purificada, por sua vez, pode ser purificada ainda mais através de resinas de troca iônica para se obter glicerina com mais de 99,5% de pureza.10
USOS TRADICIONAIS DO GLICEROL
Como podemos observar na Figura 3, o glicerol tem inúmeras aplicações na indústria,14 no entanto, a quantidade utilizada (Tabela 2)15 é muito menor do que a indústria do biodiesel produz atualmente e que ainda será produzido se, de fato, houver a implantação do B100.
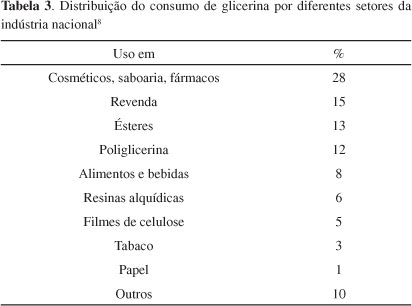
A Tabela 3 mostra o consumo industrial de glicerol no Brasil. Como principais consumidores destacam-se as indústrias de cosméticos, saboarias e fármacos, onde o glicerol é usado na forma bruta, principalmente como umectante. A utilização de derivados de transformações químicas do glicerol é ainda muito modesta.8
Alguns artigos de revisão mais recentes,1,8,14 incluindo um livro,10 abordam o uso e potenciais aplicações industriais do glicerol como matéria-prima básica da indústria química. Neste artigo de revisão, abordaremos principalmente o uso do glicerol na obtenção de blocos de construção quirais (chiral building blocks) para o emprego desses derivados na síntese de fármacos e produtos naturais.
GLICEROL EM SÍNTESES ESTEREOSSELETIVAS
Derivados do glicerol ativos opticamente são materiais de partida para síntese de compostos orgânicos importantes, tais como glicerofosfolipídeos, beta-bloqueadores, prostaglandinas, PAF (fator de agregação plaquetária) e muitos outros.19 A diferenciação entre os dois grupos enantiotópicos da molécula pró-quiral do glicerol pode de fato levar à obtenção de moléculas quirais, as quais podem ser transformadas em ambos os enantiômeros de derivados do glicerol por manipulação seletiva de um grupo funcional. Métodos de dessimetrização têm sido muito explorados, tanto na catálise química como na biocatálise, e oferecem uma tática valiosa para indução assimétrica. Em processos biocatalíticos é frequente o uso de enzimas isoladas ou de células integrais de micro-organismos.20 A resolução cinética de um racemato constitui-se também um método largamente usado para obter compostos enantiomericamente enriquecidos.21,22 A Figura 4 exemplifica, de forma geral, esses métodos para se obter compostos enantiomericamente puros ou enriquecidos, a partir do glicerol.
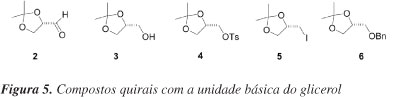
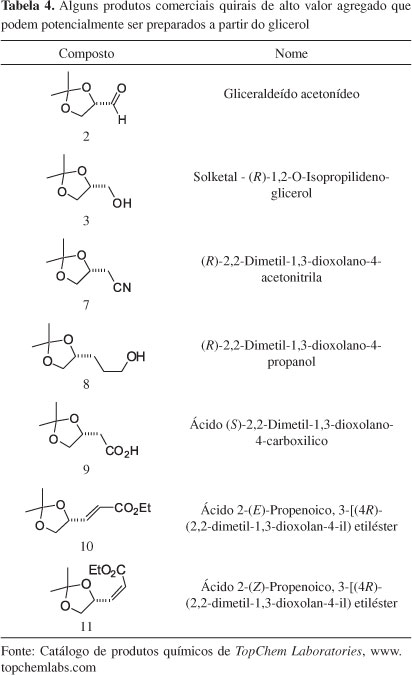
Os compostos quirais 2-6 (Figura 5), por exemplo, poderiam ser derivados do glicerol e são importantes blocos quirais com três carbonos para a síntese de moléculas enantiomericamente puras com atividades biológicas.23 Tradicionalmente, gliceraldeído acetonídeo (S)-2 é preparado a partir do D-manitol.24,25 O D-manitol é obtido industrialmente de uma mistura D-Sorbitol/D-manitol, a qual é obtida a partir de hidrólise ácida de D-Sacarose, seguida de hidrogenação catalítica.25 Isopropilideno quiral 3 e seus derivados 4-6 são obtidos pela redução de 2, e posterior manipulação do grupo hidroxila livre de 3. Esta série de compostos com configuração oposta é obtida a partir de L-serina ou ácido ascórbico.26 No entanto, os procedimentos empregados requerem o uso de tetracetato de chumbo, que é um procedimento laborioso em produção de larga escala.23
Numa rápida consulta em catálogos disponíveis na web podem-se verificar inúmeros compostos quirais que possuem a unidade básica do glicerol. Por exemplo, na Tabela 4, estão apresentados alguns compostos comerciais, obtidos de outras fontes, os quais hipoteticamente poderiam também ser preparados a partir do glicerol.
A partir do momento em que o primeiro centro quiral é produzido no glicerol, as etapas seguintes são de natureza diastereosseletiva. Estes processos são efetivados por indutores assimétricos que podem estar quimicamente ligados ao substrato (auxiliares quirais), ou simplesmente presentes no meio reagente (catálise), sendo, nos dois casos, recuperados ao final do processo. Quando se usa um auxiliar quiral covalentemente ligado ao substrato, o processo inicialmente diastereosseletivo torna-se enantiosseletivo quando o auxiliar é removido. Mesmo quando o indutor assimétrico está presente no meio reagente em quantidades catalíticas e sem estar covalentemente ligado ao substrato, usualmente se complexa com ele ativando-o para uma reação estereosseletiva. No complexo formado as faces pró-quirais do substrato são de natureza diastereotópica, as quais podem ser diferenciadas por um reagente simétrico. Com a remoção do auxiliar quiral, durante o processo de isolamento, obtém-se como produto um enantiômero puro ou uma mistura apresentando excesso enantiomérico, caracterizando-se como um processo enantiosseletivo.27
A resolução cinética, por sua vez, é geralmente definida como um processo onde um dos enantiômeros sofre uma reação química ou enzimática de menor energia de ativação, ou seja, os dois enantiômeros do racemato são transformados em produtos com velocidades de reação diferentes. Assim, em uma resolução eficiente, apenas um dos enantiômeros do racemato é seletivamente transformado em produto. Normalmente, as resoluções cinéticas reportadas, envolvendo o uso de enzimas, são altamente estereosseletivas, em condições brandas e pH neutros, produzindo excessos enantiométicos (e.e.) acima de 90%. Uma grande vantagem no uso da biocatálise está no fato de que há grande disponibilidade de enzimas no mercado, com bons preços e muitas destas enzimas podem ser empregadas na presença de solventes orgânicos.27
Catálise química
Até 1984 o glicerol nunca tinha sido focalizado como uma fonte alternativa para a preparação de síntons (blocos de construção) quirais de interesses industrial e acadêmico; nesse ano, Mukaiama et al.28 publicaram um trabalho relatando a exploração do método de resolução cinética para obter (S)-isopropilidenoglicerol (3), conhecido também comercialmente como solketal. O método envolve o tratamento do glicerol com óxido de dibutilestanho para formar o composto cíclico 12, o qual após tratamento com um cloreto de ácido puro opticamente (13) fornece dois diastereoisômeros em proporções diferentes, dependendo do cloreto de ácido usado. Após a triagem de vários reagentes acilantes, o cloreto do ácido (-)-canfâmico (13) foi o que forneceu melhor resultado, onde a estrutura cíclica e rígida dessa molécula mostrou ser importante, fornecendo dois estereoisômeros na proporção de 95:5 (Esquema 2). Entretanto, isso requer excesso de 4 mols do éster dibutilestanho, preparado a partir da reação do glicerol com óxido de dibutilestanho, para a reação subsequente obter melhor resultado.
Anos mais tarde, Hsu et al.29 exploram o uso de derivados do ácido (+)-canforssulfônico como auxiliares quirais para prepararem os blocos de construção quiral (2S)-1-benziloxi-2,3-propandiol (21) e (2R)-1-amino-2,3-propanodiol (19), como apresentado no Esquema 3. Quando uma solução de benzeno contendo 14a, glicerol e quantidade catalítica de p-TsOH é aquecida até refluxo por 36 h, com remoção de água, obtém-se o cetal 15a em 73% de rendimento. A formação do cetal 15a provavelmente acontece a partir do ataque do álcool primário Pró-R do glicerol ao grupo carbonílico na posição endo, seguida de uma ciclização com a hidroxila secundária remanescente do glicerol, a partir da posição endo, sendo todo o processo dirigido pela ligação de hidrogênio entre a hidroxila primária livre e o grupo sulfonamida adjacente. O tratamento de 15a com hidreto de sódio (1 eq.) em THF, seguido por brometo benzílico (1,2 eq.) resulta no éter benzílico 20 (89%) e subsequente hidrólise de 20 em metanol com ácido clorídrico 2 N fornece o (S)-1-benziloxi-2,3- propanodiol (21, 93%) e o auxiliar quiral 14a é recuperado completamente. A rotação específica [α]D para 21 é de -5,42º. Portanto, a configuração absoluta de C2 em 21 é S. Assim, as atribuições estereoquímicas para os compostos 15a e 20 são confirmadas por esta correlação química. Por outro lado, tratamento de 15a com cloreto metanossulfonílico e trietilamina em diclorometano, por 8 h, fornece o mesilato correspondente 16a, o qual é transformado na azida 17 (87%) que, por sua vez, é hidrolisada em metanol com HCl 2 N, fornecendo o azidodiol 18 (95%) e 14a recuperado quase quantitativamente. A azida 18 é transformada na amina correspondente 19 por hidrogenação catalítica (80%). O [α]D para 19 é de +2,96º, portanto, C2 desse composto possui configuração R.
Os rendimentos globais para os compostos 19 e 21 foram de 48 e 61%, respectivamente.
Buser e Spindler30 usaram a estratégia acima para preparar o composto 22 (um intermediário-chave na síntese de uma substância usada no controle de insetos), a partir da reação do mesilato 16b com derivados fenólicos, obtendo-se 63% de rendimento, com excesso enantiomérico (e.e.) > 98%. Por outro lado, Carlsen e Aase,31 pelo tratamento do mesilato 16a (Esquema 3) com α-naftol na presença de K2CO3 em DMSO, seguido por metanólise, produziram o composto 23, um precursor do (S)-propranolol, em 61% de rendimento (Figura 6).
Já neste século, Marzi et al.32 usaram também essa mesma estratégia para sintetizar (R)-carnitina (31) a partir do glicerol, usando a canforsulfonamida 24. O glicerol é primeiramente dessimetrizado, de acordo com o procedimento descrito por Hsu et al.,29 fazendo uso de um derivado da (1R)-(-)-10-canforsulfonamida 24 como auxiliar quiral. A estereoquímica da canforsulfonamida é escolhida com base na quiralidade do isômero da carnitina a ser sintetizado. A informação quiral é introduzida no glicerol por reação com um dos derivados da sulfonamida selecionado (24c). O processo global é descrito no Esquema 4.
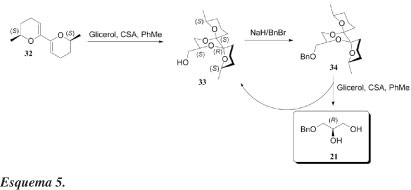
Somente um dos spiro-acetais, 25c, dos quatro estereoisômeros possíveis, é formado, com 60% de rendimento. A configuração correta S do derivado do glicerol 25c é estabelecida com base na conversão final para (R)-(-)-carnitina (31). O álcool 25c é então convertido, com rendimento quase quantitativo, no seu mesilato correspondente 26c e o novo grupo de saída criado é finalmente deslocado pela trietilamina para formar 27, carregando o grupamento trietilamônio, característico da carnitina. O auxiliar quiral é então removido com HCl e posteriormente recuperado da fase orgânica após elaboração da mistura reacional, enquanto o mesilato 28 foi obtido por evaporação da solução aquosa. Após mais 4 etapas (R)-(-)-carnitina 31 é obtida. O rendimento global, partindo do glicerol, é de 40% e o produto final foi com excesso enantiomérico superior a 98%.Em outra abordagem interessante, Boons et al.33 descreveram um processo que realiza simultaneamente proteção e dessimetrização enantiosseletiva do glicerol. Este processo é ilustrado pela reação do glicerol com bis-dimetil-di-hidropirano 32 (Esquema 5). O trabalho possibilitou a preparação de um novo equivalente sintético (21) do gliceraldeído acetonídeo (2), o qual é um bloco de construção quiral importante em síntese orgânica.34
A reação de 32 com glicerol, na presença do catalisador ácido canforssulfônico (CSA) em ebulição com tolueno, resulta numa completa diastereosseletividade, para dar o dispirocetal 33. A configuração absoluta de 33 pôde ser determinada por dados espectrais de RMN de seu éster de Mosher correspondente. O álcool 33 é convertido no éter benzílico correspondente 34, por tratamento com hidreto de sódio e brometo de benzila. Tratamento de 34 com glicerol e ácido canforssulfônico fornece (R)-1-O-benzil glicerol 21, juntamente com o dispirocetal 33, o qual é recuperado. O composto 33 é então obtido como um único diastereoisômero, fornecendo, portanto, um processo extremamente eficiente de reciclagem. Assim, durante a etapa de desproteção, o grupo protetor quiral é preservado. Nesse trabalho, o composto (R)-21 foi convertido em derivados análogos do isopropilideno.
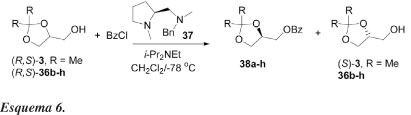
Meia década atrás, Terakado e colaboradores35 relataram o primeiro exemplo de resolução cinética organocatalítica de alcoóis primários derivados do glicerol, fazendo uso de 1,2-diaminas derivadas de (S)-prolina (Esquema 6).
Normalmente, este tipo de catálise é feita com enzimas, obtendo-se altas seletividades. Os autores investigaram a resolução cinética de derivados do glicerol contendo duas hidroxilas protegidas, como um substrato modelo para alcoóis primários racêmicos, baseados no trabalho pioneiro de Mukaiyama e colaboradores.28 O tratamentdo de dois alcoóis (R,S)-3 e 36b-h com cloreto de benzoíla, sob a influência de (S)-1-metil-2-[(benzilmetilamino)metil]pirrolidina (37), combinado com di-isopropiletilamina a -78 ºC, em diclorometano, resulta nos benzoatos correspondentes 38a-h, com rendimentos entre 44-54% e e.e. entre 23-60%. Os alcoóis não reagidos são isolados com rendimentos entre 33-47% e e.e. entre 29-82% (Esquema 6, Tabela 5). A configuração absoluta do éster 6 é assinalada como sendo R, por comparação da rotação óptica registrada na literatura. Substratos substituídos com n-Bu ou ciclo-hexilideno mostraram seletividade moderada. Quando 1,3-dioxolanos impedidos estericamente com dois grupos isopropila foram usados (exp. 5, Tabela 5), obtêm-se altos valores de enantiosseletividade (E = 7,3). 1,3-Dioxolanos contendo grupos ciclo-hexila e fenila tiveram resultados comparáveis do ponto de vista de seletividade. A enantiosseletividade foi menor quando se usou grupos benzila.
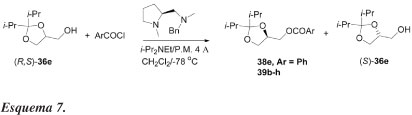
Terakado e colaboradores35 avaliaram também o efeito do agente acilante na resolução cinética do álcool 36e (Esquema 7, Tabela 6). Benzoatos com substituintes em para são mais seletivos que aqueles com substituintes em orto e meta. Entre os p-substituintes testados, tais como grupos retiradores ou doadores de elétrons (experimentos 4-7, Tabela 6), o grupo metil conduz a uma enatiosseletividade E = 9,8 (exp. 4). Com o cloreto de α-naftoila obtém-se baixa seletividade e baixo rendimento químico (exp. 8) devido à sua insolubilidade em diclorometano a -78 ºC. No entanto, quando uma mistura de solventes [CH2Cl2-DMF (9:1)] é usada, alta seletividade (E = 16) é alcançada (exp. 9).
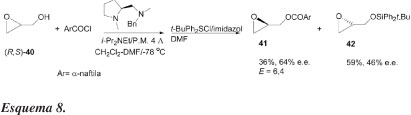
Nesse trabalho, são também estudadas as reações de vários racematos de alcoóis primários heterocíclicos, entre eles o glicidol (40). A resolução cinética do racemato de 40 foi realizada por acilação, seguida de sililação do álcool não acilado. Dessa maneira, o benzoato (41) e o éter de silício (42) correspondentes são obtidos em 36% de rendimento e 64% de e.e. e 59% de rendimento e 46% de e.e., respectivamente (Esquema 8).
O glicidol (40) opticamente ativo e seus derivados são considerados compostos versáteis para serem empregados em sínteses quirais. Nesse sentido, ambos os enantiômeros do glicidol se tornaram amplamente utilizados como matérias-primas para a síntese de muitos compostos interessantes, tais como drogas anticâncer, inibidores da síntese de proteínas, bem como um derivado de 2-oxazolidinona usado contra a depressão.36
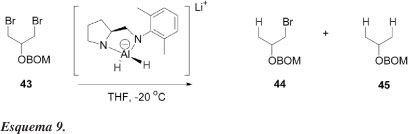
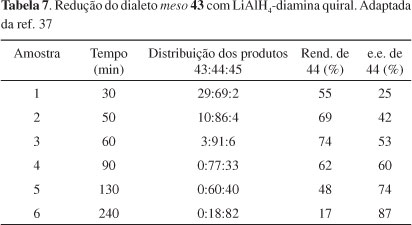
Chong et al.37 relataram a redução seletiva de meso 1,3-di-haletos obtidos do glicerol, usando-se LiAlH4-diamina como agente redutor quiral, com moderados níveis de diferenciação enantiotópicas (Esquema 9). As reações foram conduzidas em THF e os dados experimentais estão apresentados na Tabela 7. Os resultados sugerem que a redução 43 → 44 ocorre muito mais rapidamente que a redução subsequente 44 → 45. Em apenas 1 h de reação (exp. 3, Tabela 7), o produto 44 já é majoritário. A seletividade inicial é modesta (exp. 1, onde pequena quantidade de 45 é formada); mas as resoluções cinéticas subsequentes de 44 aumentam sua pureza óptica e, após um período prolongado de reação, 44 pode ser isolado com razoável pureza enantiomérica, mas com baixo rendimento químico. Apesar disso, esses resultados mostraram pela primeira vez que a dessimetrização enantiosseletiva de di-haletos meso com agentes quirais é perfeitamente plausível.
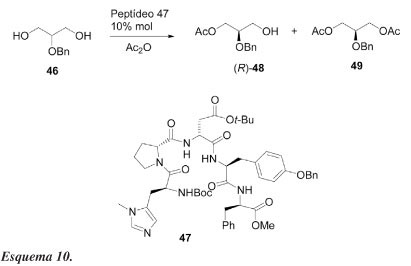
Lewis et al.38 demonstraram que um catalisador pentapeptídeo pode levar à dessimetrização efetiva de um derivado simples do glicerol e com enantiosseletividade comparável à exibida por lipase de Pseudomonas sp. Quando o derivado do glicerol 46 é solubilizado em PhCH3/CH2Cl2 (12:1) a -55 ºC e exposto ao anidrido acético e ao peptídeo 47 (10 mol%), ocorre acilação enantiosseletiva para formar (R)-48 (Esquema 10). As quantidades relativas do monoacetato 48 e do diacetato 49 podem ser reguladas como uma função estequiométrica do anidrido. Como mostrado na Tabela 8 (exp. 1), quando 1,5 eq. de anidrido acético são utilizados, (R)-48 é obtido com 52% de rendimento e 86% de e.e. Quando a quantidade de anidrido é aumentada, o excesso enantiomérico global do monoacetato é também aumentado, no entanto, o rendimento químico diminui.
Catálise enzimática
Para as sínteses de derivados quirais do glicerol, abordagens enzimáticas, tais como resolução cinética de racematos ou dessimetrização do glicerol e seus derivados simétricos pró-quirais, têm sido desenvolvidas. Esses métodos são baseados na habilidade principalmente de lipases em distinguir entre grupos enantioméricos (resolução cinética) e enantiotópicos (dessimetrização) e usualmente são empregados solventes orgânicos como meio.39
A seguir, apresentaremos os principais trabalhos desenvolvidos usando-se enzimas como catalisadores na preparação de derivados quirais do glicerol. Com o intuito de facilitar a leitura, os trabalhos foram organizados em dois grupos: dessimetrização e resolução cinética.
Dessimetrização
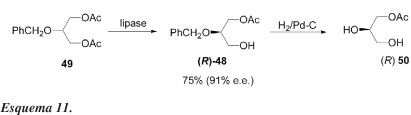
Em 1986, Breitgoff e colaboradores40 descreveram o primeiro sucesso na obtenção de blocos quirais de construção a partir do glicerol, utilizando catálise enzimática. Estudaram a reação de hidrólise enzimática de 49 (Esquema 11) na presença de várias lipases. Os resultados estão sumariados na Tabela 9.
Idealmente, em uma transformação enantiosseletiva alta, somente um grupo acetato deveria ser removido a fim de se obter altos rendimentos químicos e ópticos. Os melhores resultados, nesse sentido, foram obtidos com lipase pancreática de porco (E.C.3.1.1.3) e com lipase lipoproteica (E.C.3.1.1.34), sendo que esta última produziu (R)-48 com bom rendimento químico (75%) e alta pureza óptica (e.e. = 91%) como determinado por espectros de RMN de 1H dos ésteres de Mosher diastereoisoméricos. Por hidrogenólise, o acetato (R)-48 é transformado no já conhecido monoacetato (R)-50 {[α]D20 = -9,2º (c 1,7 piridina)}. Dessa forma, confirma-se tanto a pureza óptica como a configuração absoluta.
O composto 48 com configuração oposta (S) e outros α-monoacilglicerois (MAGs) podem ser obtidos por transesterificação ou esterificação, catalisadas por lipases em solventes orgânicos.41,42
Murata et al.42 relataram a preparação do composto (S)-48 a partir de derivados do glicerol 2-O-substituídos. Inicialmente, 2-O-benzilglicerol 46 foi esterificado enzimaticamente com ácido acético em um solvente orgânico (Esquema 12). Em todos os casos, conforme mostrado na Tabela 10, as lipases testadas catalisaram enantiotopicamente as esterificações para fornecer o composto (S)-48 com rendimentos ópticos variando de moderados a bons. Esses resultados serviram para indicar que esterificação catalisada por lipases procederia em meio orgânico e a enzima poderia ser removida facilmente por simples filtração.
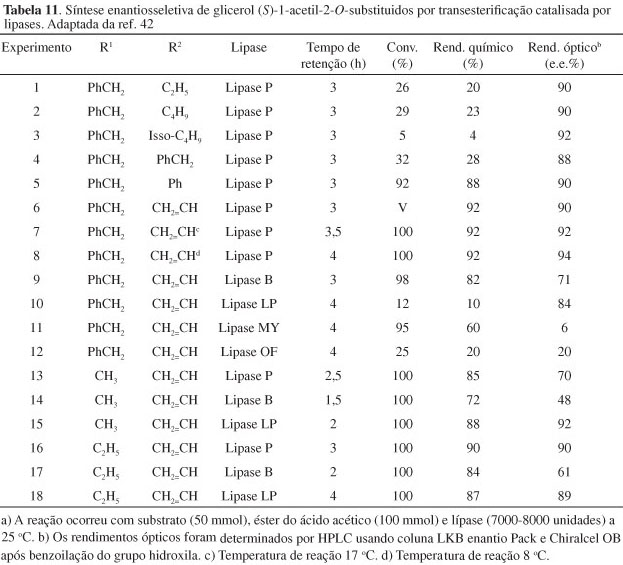
No trabalho de Murata foi verificado que a transesterificação de um derivado do glicerol por um éster carboxílico, catalisada por lipase, poderia ser feita em um sistema onde o agente de transesterificação fosse o próprio solvente da reação. Por exemplo, quando a mistura de 2-O-benzilglicerol, acetato de etila e lipase P é agitada vigorosamente a 25 ºC por 3 h, (S)-1-O-acetil-2-O-benzil-glicerol é obtido com 20% de rendimento e pureza óptica de 90% (Esquema 13, Tabela 11). Nesse estudo, os autores obtêm resultados excelentes com reação de transesterificação entre derivados 2-O-substituídos e ésteres carboxílicos, catalisada por diversas lipases. Em particular, transesterificação com acetato de vinila ou acetato de fenila fornece altos rendimentos, tanto químico como óptico (Tabela 11).
Dessimetrização da molécula do glicerol usando catálise enzimática ou química é um processo atraente, pois em um único evento pode-se obter derivados opticamente ativos diretamente sem nenhuma modificação prévia. Como exemplo, para esses derivados importantes, pode-se citar os α-monoacilglicerois opticamente ativos (MAGs), os quais são usados como material de partida para a síntese de drogas quirais, tais como β-bloqueadores, fator de ativação de plaquetas (PAF), (S)-carnitina e γ-amino-β-hidroxibutírico (GABOB).39,43
Os MAGs são comumente preparados por síntese química catalítica, no entanto, atualmente há um grande interesse para o uso de métodos quimio-enzimáticos para suas sínteses.39 Nessa direção, Kato et al.44 reportaram um método para sintetizar o monobenzoilglicerol 56 opticamente ativo, via transesterificação enzimática do glicerol com benzoato de vinila (55) em 1,4-dioxano (Esquema 14).
Usando Chirazyme L-2 (lipase de Candida antartica) (R)-56 é preparado em uma única etapa, com excesso enantiomérico >95%. Após extração e purificação em coluna cromatográfica de sílica gel, o produto da reação é obtido com rendimento de 94% e sua estrutura confirmada como sendo (R)-56 (54% e.e.) por RMN, IR, espectrometria de massas e CLAE quiral. Sua pureza óptica pode ser elevada para mais de 95%, por recristalização com hexano/iPrOH.
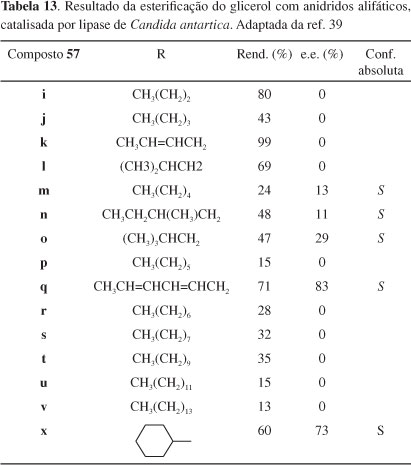
Esterificação seletiva do glicerol com anidridos aromáticos e alifáticos, catalisada por também por Chirazyme L-2, foi efetuada por Batovska e coautores.39 A assimetrização mediada pela lipase de Candida antartica da molécula pró-quiral do glicerol é conseguida via acilação dos grupos hidroxila primários, em dioxano, como mostrado no Esquema 15. Os resultados estão mostrados nas Tabelas 12 e 13.
Como se pode ver, os agentes acilantes aromáticos possuem um substituinte na posição para e um ou dois grupos metileno entre o anel aromático e o grupo acila; os anidridos alifáticos possuem cadeias lineares ou ramificadas variando de 3 a 14 carbonos. Interessantemente, a lipase usada mostrou seletividade pró-R para a esterificação com anidridos aromáticos e pró-S ou nenhuma seletividade para os agentes acilantes alifáticos. A introdução de ligações duplas conjugadas no agente acilante aumentou drasticamente a seletividade enantiotópica da enzima. Em comparação com anidridos alifáticos, os agentes acilantes aromáticos esterificaram o glicerol muito mais lentamente. Está evidente, portanto, que efeitos estéricos e eletrônicos dos grupamentos anidridos afetam a eficiência da lipase.
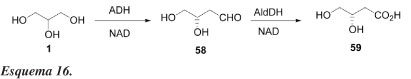
Existem outros métodos para se obter compostos úteis diretamente do glicerol. Como exemplo, bio-oxidação enantiosseletiva de glicerol para ácido L-glicérico (59), usando as enzimas álcool e aldeído desidrogenases como catalisadores, foi realizada com sucesso por Wong e Matos.45 Como indicado no Esquema 16, a enzima álcool desidrogenase de fígado de cavalo (HLADH) catalisa seletivamente a transformação do glicerol para L-gliceraldeído (58) que pode ser posteriormente convertido para ácido L-glicérico (59) pela enzima aldeído desidrogenase (AldDH).
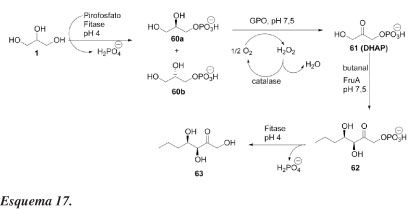
Schoevaart et al.46 publicaram um procedimento one-pot muito interessante, envolvendo cascata de 4 etapas enzimáticas, para a síntese de carboidratos a partir do glicerol. Conforme ilustrado no Esquema 17, o glicerol é tratado com pirofosfato, na presença de fitase, fornecendo o racemato de glicerol-3-fosfato (60a e 60b) com rendimento quantitativo. Seletivamente, o L-glicerol-3-fosfato (60b) é oxidado a fosfato de di-hidroxiacetona (DHAP, 61), em pH 7,5 na presença de glicerolfosfato oxidase (GPO) e catalase. Subsequentemente, a enzima frutose-1,6-bisfosfato aldolase foi usada para catalisar a reação aldólica entre DHAP e butanal formando-se o composto 62. Desfosforilação do aduto aldólico 62 foi catalisado por fitase em pH 4,0, fornecendo 5-desoxi-5-etil-D-xilose (63) em rendimento de 57% a partir de L-glicerol-3-fosfato. A manipulação do pH permitiu total controle sobre a atividade das fitases, a qual pode ser desativada aumentado-se o pH de 4,0 para 7,5 e ativada abaixando o pH novamente para 4,0. Esta combinação de quatro enzimas diferentes em transformações enzimáticas one-pot em cascata proporciona um procedimento atrativo para realizar reações aldólicas com DHAP aldolases, usando-se materiais de partida baratos e facilmente disponíveis, como glicerol e pirofosfato.
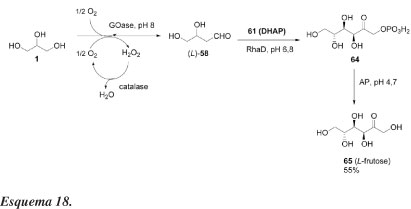
Ainda visando a síntese de açúcares, Franke et al.47 relataram outro processo enzimático interessante para sintetizar L-monossacarídeos a partir de oxidação seletiva do glicerol. A combinação de glicerol, GOase, catalase, DHAP (61) e RhaD resulta na formação de L-frutose-1-fosfato (64) como um único produto (comprovado por espectros de RMN de 1H). A desfosforilação de 64 é efetuada com ácido fosfatase (AP) na presença das outras enzimas sem prévio isolamento (Esquema 18).
Segundo os autores, L-gliceraldeído (58) é estável na mistura reacional aquosa em pH 6,7, mesmo quando aquecida a 100 ºC. Esse método fornece L-frutose com alta pureza óptica e com rendimento químico bom (55%), constituindo um processo eficiente e ambientalmente adequado. Monossacarídeos não naturais têm inúmeras aplicações. L-frutose, por exemplo, é conhecida como um edulcorante não nutritivo, um inibidor de várias glicosidases e um inseticida para formigas. Além disso, monossacarídeos não naturais são potencialmente usados como blocos de construção quiral para a síntese de compostos ativos biologicamente.
Resolução cinética
Em geral, a resolução cinética é feita usando-se alcoóis primários derivados do glicerol ou por hidrólise de ésteres desses alcoóis.
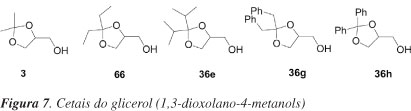
O primeiro método de resolução enzimática de alcoóis primários, oriundos do glicerol, foi apresentado por Terao et al..48 Os autores relataram um processo para resolução de cetais do glicerol (1,3-dioxolano-4-metanol). A esterificação de racematos dos alcoóis, mostrados na Figura 7, com anidrido succínico e catalisados por lipases leva à formação dos monoésteres succínicos correspondentes de forma enantiosseletiva, os quais são separados facilmente dos alcoóis que não reagiram após tratamento da mistura com uma solução alcalina. A reação é efetuada pela incubação, a 25 ºC, de uma mistura do cetal (5 mmol), anidrido succínico (5 mmol) e lipase P de Pseudomonas fluorescens ou lipase B de Psedomonas fragi (1.600 un), em éter etílico. Após remoção da lipase por filtração, a fase etérea é lavada com solução de Na2CO3 1 mol L-1. Um dos enantiômeros é obtido a partir da fase etérea e o outro após tratamento da fase aquosa com NaOH 10%. Substituintes na posição 2 do cetal afetam a enantiosseletividade (experimentos 1-5, Tabela 14). O grupo di-isopropila fornece bom resultado com o uso de lipase P (exp. 3). O melhor resultado com respeito à enantiosseletividade é obtido usando lipase B. O valor do excesso enantiomérico (e.e.) do éster isolado (rendimento de 41%) é de 92% (exp. 5).
Por outro lado, hidrólises enantiosseletivas de ésteres de 1,3-dioxolano-4-metanol foram investigadas por Partali et. al.,49 usando lipases e proteinases. Os autores estudaram a influência do grupo cetal e da parte acíclica nessas hidrólises. Várias lipases e proteinases foram testadas e a melhor enantiosseletividade (E) foi obtida com proteinase de Aspergillus oryzae, a qual forneceu um valor de E = 9 com butanoato de 2,2-dimetil-1,3-dioxolano-4-metanoila, fornecendo o solketal (3) com configuração S.
A enantiosseletividade, expressa pela razão enantiomérica (E), é o índice mais importante para resoluções químicas ou enzimáticas de racematos: um valor alto de E indica um alto valor de excesso enantiomérico (e.e.) combinado com um alto rendimento químico.50
Posteriormente, Vänttinen e Kanerva51 relataram que resolução catalisada por lipase de Psedomonas flurorescens nas transesterificação do solketal racêmico com 2,2,2-trifluroetilcarboxilatos (principalmente hexanoato, butirato e 3-metilbutirato) dá valores moderados de E: 16 a 23 ºC e 27 a 0 ºC. Metanólise enzimática de ésteres do solketal racêmico procede com E = 24-26.
Para aumentar e eficiência da enantiosseletividade, os autores usaram resolução cinética dupla e, tanto a sequência acilação enzimática-saponificação química-acilação enzimática-saponificação química (Esquema 19) ou acilação química-alcoólise enzimática-acilação (Esquema 20) produzem bons resultados; 97% de e.e. para butirato (R)-67 e 98% para hexanoato (S)-68.
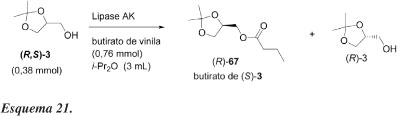
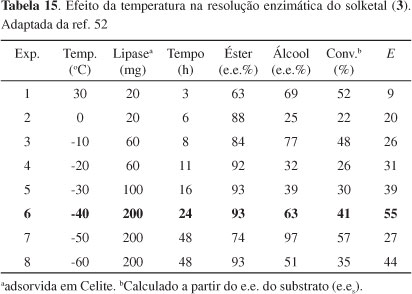
Sakai et al.52 estudaram o efeito da temperatura na resolução cinética de (R,S)-3 catalisada por lipase AK de Pseudomonas fluorences e conseguiram aumentar o valor de E de 9 (a 30 ºC) para 55 (a -40 ºC) em éter isopropílico, com e.e. de 93%, apesar da necessidade de grande quantidade de lipase e longo tempo de reação. Solketal racêmico (3) foi tratado com butirato de vinila em éter isopropílico na presença da lipase AK (Esquema 21). O resultado dessas reações, usando-se enol-éster, está mostrado na Tabela 15.
A exemplo do grupo de Sakai, Miyazawa et al.53 também fizeram uso de transesterificação irreversível catalisada por lipase, para resolver dois cetais do glicerol (Esquema 22).
Inicialmente, os autores testaram a resolução enzimática do solketal (3) via acilação com acetato de vinila em éter isopropílico. Foram usadas cerca de 12 lipases de fontes microbianas e pancreática, no entanto, somente os resultados com 3 lipases foram apresentados no artigo para a resolução do solketal. A enantiosseletividade, incluindo a preferência estereoquímica (R ou S), variou consideravelmente com a enzima usada. Nenhuma das lipases testadas mostra alta enantiosseletividade, exceto a lipase de Rhisopus javanicus, a qual apresenta grau moderado de enantiosseletividade (E = 11, 48% de e.e. para o enantiômero S), mas com tempo de reação muito longo (280 h). Por outro lado, resolução enzimática de 2,2-difenil-1,3-dioxolano-4-metanol (36h) com butanoato de vinila fornece melhores resultados. O valor de E é calculado a partir da conversão determinada por CLAE e os excessos enantioméricos são determinados por CLAE com adsorvente quiral. A Tabela 16 apresenta os resultados da transesterificação do álcool 36h, usando várias lipases de diferentes fontes. As lipases do gênero Rhizopus mostram maiores enantiosseletividades comparada com as de outras fontes. Lipase de Rhizopus delemar apresenta maior enantiosseletividade (E = 23). Entre os solventes testados nessas reações, éter isopropílico é melhor. Uma resolução de escala multigramas produz o (R)-álcool com 95% de e.e. e 33% de rendimento.
Transesterificação catalisada por lipase de 2,2-difenil-1,3-dioxolano-4-metanol substituídos foi investigada por Kawanami et al..43 Nesse trabalho, os autores descrevem o estudo do efeito do substituinte sobre a enantiosseletividade na resolução cinética de vários 1,2-cetais do glicerol com difenilas substituídas (70b-f), bem como efeitos do solvente e temperatura. Os cetais 36h e 70b-f são preparados pela reação do glicerol com a benzofenona 4-substituída correspondente, na presença de ácido p-toluenossulfônico (Esquema 23). Para investigar o efeito do solvente, o composto 70e é selecionado como substrato modelo e a transesterificação, catalisada pelas lipases OS (PCL) e AK (PFL), é realizada em vários solvente a 25 ºC. A configuração absoluta do enantiômero majoritário do álcool 70e é determinada como sendo R, por comparação com as rotações ópticas.
A acilação do cetal 70e com butirato de vinila lipase AK em i-Pr2O é um pouco mais enantiosseletiva (E =11) do que a acilação do solketal realizada por Sakai (E = 9),52 sob condições reacionais similares. Uso de acetato de vinila como agente acilante aumenta discretamente a enantiosseletividade (E = 18). Hexano seco é melhor solvente (E = 18) do que hexano saturado com água na resolução cinética do substrato 70e usando-se lipase AK. No caso da lipase PS, efeito similar é observado e hexano seco também prova ser o melhor solvente (E = 21) entre todos os testados (Tabela 17).
Os autores examinaram o efeito do substituinte nos grupos fenila usando os substratos 36h e 70b-f e acetato de vinila em hexano seco a 25 ºC. Como se pode observar na Tabela 18, a enantiosseletividade da lipase AK para esses cetais diminui com o aumento do tamanho do grupo doador de elétrons (exp. 1-3), e para os substratos 70d-f os valores de E aumentam com o tamanho dos grupos halogênios, retiradores de elétrons, sobre o anel aromático. Por outro lado, a enatiosseletividade da lipase PS aumentou para os substratos 36h, 70b-c e 70d-f com o aumento do tamanho de ambos os substituintes (experimentos 7-12). O composto 70f (R = Br) teve enantiosseletividade mais alta (E = 36).
O efeito da temperatura nessas reações foi estudado usando a lipase PS em hexano seco a 0 ºC. Os valores de E para os substratos 36h, 70b e 70d (R = H, Me, F) não sofreram nenhuma mudança a 0 ºC. No entanto, os valores de E para os compostos 70e e 70f (R = Cl, Br) aumentaram de 21 para 25 e de 36 para 57, respectivamente. Por outro lado, E diminuiu para o substrato 70c (R = OMe).
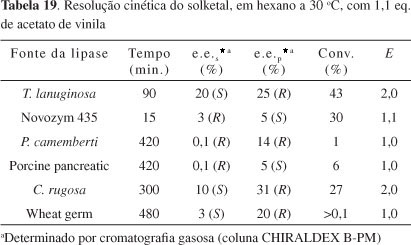
Araújo et al.,19 em estudos preliminares, testaram algumas lipases que não tinham sido utilizadas anteriormente para resolver eficientemente o solketal. (R,S)-3 é submetido à resolução cinética em hexano anidro por 6 lipases de fontes diferentes e acetato de vinila foi usado como agente acilante. De acordo com a Tabela 19, todas as 6 lipases mostram baixos valores de E, usando 1,1 equivalentes do agente acilante (30 ºC). Interessantemente, nessa comunicação curta, é relatado que quando se usam 2,2 equivalentes de acetato de vinila a conversão é baixa, mas é observado um grande aumento no excesso enantiomérico (e.e.>99%), promovido pela lipase de P. camemberti, o que demonstra ser promissor estudar o uso dessa lipase na resolução enzimática do solketal.
Além dos cetais, 2,3-carbonato de glicerol tem sido usado para preparar blocos de construção quiral importantes para a síntese orgânica. 2,3-Carbonato de glicerol (72) é um composto estável, líquido incolor que pode ser usado em diversas aplicações, incluindo a síntese de compostos enantiomericamente puros de grande interesse biológico.53 Algumas rotas sintéticas são descritas na literatura para preparar este derivado, incluindo carboxilação do glicerol com dióxido de carbono, catalisado por Sn,54 zeólitas ou resinas de trocas iônicas.55 Recentemente, Mota e co-laboradores8 desenvolveram um método para produzir o carbonato de glicerol, reagindo a bioglicerina bruta, proveniente da produção de biodiesel sem qualquer purificação, com N,N'-carbonil-di-imidazol (CDI). Adicionalmente, a síntese enzimática do carbonato de glicerol, a partir de glicerol e dimetil carbonato, foi efetuada com sucesso por Kim et al.53 usando lipase imobilizada de Candida antarctica (CALB, Novozym 435) como catalisador. O uso equimolar de glicerol e carbonato de dimetila na reação catalisada por Novozym 435 forneceu carbonato de glicerol com rendimento quase quantitativo. O produto resultante dessa reação, no entanto, mostrou baixo excesso enantiomérico (13%) do enantiômero de configuração R.
Pallavicini et al.56 relataram a resolução cinética de 2,3-carbonato glicerol (72), via transesterificação catalisada por lipase de Pseudomonas fluorescens (PFL) do racemato e subsequente alcoólise do acetato 73, enriquecido opticamente do enantiômero S na presença da mesma enzima. As duas reações subsequentes, feitas em clorofórmio, forneceram (R) e (S) 2,3-carbonato glicerol, com altos rendimentos químicos e ópticos, respectivamente (Esquema 24).
Nesse trabalho foi avaliado o efeito de quatro solventes na enantiosseletividade do processo (álcool terc-amílico, THF, éter di-isopropílico e clorofórmio). Somente clorofórmio apresenta resultados excelentes (E = 16). Os autores sugerem que a influência favorável do CHCl3 pode ser devida à sua propriedade peculiar de alta hidrofobicidade combinada com boa polaridade.
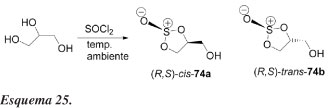
Lemaire e Bolte57 relataram a primeira resolução cinética catalisada por lipase de um sulfito do glicerol. Os autores mostram que cis-2-oxo-5-hidroximetil-1,3,2-dioxatiolano 74a racêmico (Esquemas 25 e 26) pode ser resolvido eficientemente por uma reação de acilação com butirato de vinila, catalisada pela lipase de Pseudomonas cepacia. A enzima é imobilizada em celite na presença de sucrose.
Nos experimentos de controle a reação não procede sem enzima. Uma vez mais, para um derivado do glicerol, a lipase de P. cepacia é selecionada para uma resolução enzimática e mostra uma preferência para o enantiômero de configuração S. Solventes halogenados, como diclorometano e clorofórmio, influenciam favoravelmente a enantiosseletividade, com relação ao isômero cis (74a). Embora alcoóis primários não sejam a situação mais favorável para a resolução enzimática, a lipase de P. cepacia catalisou a acilação enantiosseletivamente (E = 26) do sulfito cíclico 74a podendo apresentar uma eficiente preparação de (S)-75a e (R)-74a.
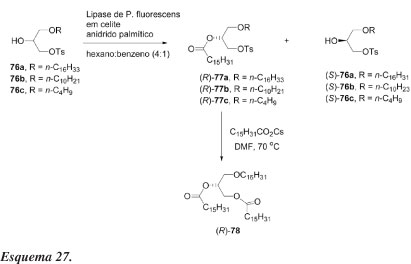
A resolução enzimática de alcoóis secundários derivados do glicerol foi efetuada por Chênevert e Gagnon58 para sintetizar (S)-(+)-1-O-hexadecil-2,3-di-O -hexadecanoilglicerol (78), um produto natural marinho. Derivados 1-O-alquil-3-O-tosilglicerol racêmicos foram resolvidos por acilação com anidrido palmítico na presença de lipase de Psedomonas fluorescens em meio orgânico. As reações são altamente seletivas e procedem sem migração de grupos, o que é reconhecido como um sério problema na química do glicerol. Substituição do tosilato por carboxilato ou fosfato pode levar a um grande número de éteres naturais.58
Os alcoóis racêmicos 76a-c (Esquema 27) são preparados a partir de solketal, por métodos bem estabelecidos. Racematos desses alcoóis e anidrido palmítico em quantidades estequiométricas são dissolvidos em hexano/benzeno 4:1. Após adição de PFL adsorvida em celite, o progresso da reação é monitorado por CLAE e interrompida quando a mesma atinge cerca de 50% de conversão. Os produtos resultantes dos experimentos são facilmente separados em coluna de cromatografia do tipo flash.
O excesso enantiomérico de (R)-77 é igual ou superior a 95% e do material de partida recuperado, (S)-76, entre 94 e 96%. A reação inversa, hidrólise enzimática do éster correspondente em éter isopropílico saturado com água, foi também altamente estereosseletiva (99% e.e.) para fornecer o álcool (R)-76.
CONCLUSÕES
Quase sempre, o uso de substratos oriundos de resíduos renováveis é uma excelente opção, ambientalmente correta, para o desenvolvimento de novos produtos de maior valor agregado, bem como contribui para a redução do custo do tratamento dos resíduos e aumenta o valor econômico desses coprodutos, com consequente diminuição do custo de produção do produto principal. O glicerol é um coproduto do biodiesel e está dentro deste contexto. Alguns commodities químicos produzidos a partir do petróleo poderiam ser obtidos a partir do glicerol, usando-se catalisadores químicos ou enzimáticos. Esta revisão procurou mostrar que o glicerol pode ser utilizado na síntese orgânica moderna como substrato de partida na síntese enantiosseletiva de blocos de construção quirais (chiral building blocks) para fármacos e produtos naturais.
Finalmente, o glicerol é uma matéria-prima barata e muito versátil, constituindo-se em uma alternativa real e viável para gerar substâncias que possam atuar como precursores de produtos industriais de alto valor agregado. O desenvolvimento de processos inteiramente "verdes" para converter facilmente o glicerol em substâncias quirais, com razão enantiomérica (E) alta, é desejável para tornar a produção do biodiesel mais econômica, promovendo o uso do biodiesel e reduzindo a dependência do petróleo. Nesse aspecto, o químico orgânico sintético tem um papel fundamental nessas transformações, seja na descoberta e desenvolvimento de agentes de indução assimétrica como auxiliares, reagentes ou catalisadores quirais, incluindo células integrais de micro-organismos e enzimas isoladas, como no desenvolvimento de métodos eficientes para o uso desses agentes, visando obter produtos econômicos, social e ambientalmente adequados.
AGRADECIMENTOS
À Fundação de Apoio ao Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT-MS) pelo apoio financeiro e bolsa de mestrado à Y. J. K. Araújo.
REFERÊNCIAS
1. Zhou, C. H.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q.; Chem. Soc. Rev. 2008, 37, 527.
2. King, A. G.; Wright, M. W.; J. Chem. Educ. 2007, 84, 202.
3. Pinto, A. C.; Guarieiro, L. N.; Rezende, M. J.; Ribeiro, N. M.; Torres, E. A.; J. Braz. Chem. Soc. 2005, 16, 1313.
4. Santos, A. P.; Pinto, A. C.; Quimica Nova na Escola 2009, nº 31, 58.
5. http://www.biodiesel.gov.br/, acessada em Junho 2009.
6. Geris, R.; Santos, N. A. C.; Amaral, B. A.; Maia, I. S.; Castro, V. D.; Carvalho, J. R. M.; Quim. Nova 2007, 30, 1369.
7. Mota, C. J. A.; Tchê Química 2006, 3, 26.
8. Mota, C. J. A.; da Silva, C. X. A.; Gonçalves, V. L. C.; Quim. Nova 2009, 32, 639.
9. http://www1.eere.energy.gov/biomass/pdfs/35523.pdf, acessada em Janeiro 2009.
10. Pagliaro, M.; Rossi, M.; The Future of Glycerol - New Usages for a Versatile Raw Material, RSC Publishing: Cambridge, 2008.
11. Katha, S. S.; Medical Hypotheses 1999, 53, 338.
12. Lide, D. R., editor-in-chief; Handbook of Chemistry and Physics, 87th ed., Taylor and Francis Group: Boca Raton, 2006.
13. Callam, C. S.; Singer, S. J.; Lowary, T. L.; Hadad, C. M.; J. Am. Chem. Soc. 2001, 123, 11743.
14. Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Pina, C. D.; Angew. Chem., Int. Ed. 2007, 46, 4434.
15. Wang, Z. X.; Zhuge, J.; Fang, H.; Prior, B. A.; Biotechnology Advances 2001, 19, 201.
16. de Arruda, P. V.; Rodrigues, R. C. L. B.; Felipe, M. G. A.; Revista Analytica 2006, 26, 56.
17. Andrews, M. A.; Klaeren, S. A.; US pat. 5,026,927 1991.
18. http://www.aboissa.com.br/, acessada em Novembro 2008.
19. Araújo, Y. J. K.; de Lima, D. P.; Beatriz, A.; Orbital - The Electronic Journal of Chemistry 2009, 1, 1.
20. Faber, K.; Biotransformation in Organic Chemistry, 4th ed., Springer: Berlin, 2000.
21. Fessner, W-D.; Anthonsen, T., eds.; Modern Biocatalysis - Stereoselective and Environmentally Friendly Reactions, Wiley-VCH: Weinheim, 2009.
22. Strübing, D.; Krumlinde, P.; Piera, J.; Bäckvall, J-E.; Adv. Synth. Catal. 2007, 349, 1577.
23. Uang, B. J.; Po, S. Y.; Hung, S. C.; Liu, H. H.; Hsu, C. Y.; Lin, Y. S.; Chang, J. W.; Pure Appl. Chem. 1997, 69, 615.
24. Debost, J. L.; Gelas, J.; Horton, D.; J. Org. Chem. 1983, 48, 1381.
25. de Oliveira, P. S. M.; Ferreira, V. F.; de Souza, M. V. N.; Quim. Nova 2009, 32, 441.
26. Jung, M. E.; Shaw, T. J.; J. Am. Chem. Soc. 1980, 102, 6304.
27. Barreiro, E. J.; Ferreira, V. F.; Costa, P. R. R.; Quim. Nova 1997, 20, 647.
28. Mukaiyama, T.; Tanabe, Y.; Shimizu, M.; Chem. Lett. 1984, 401.
29. Hsu, C. Y.; Lin, Y. S.; Uang, B. J.; Tetrahedron: Asymmetry 1990, 1, 219.
30. Buser, H. P.; Spindler, F.; Tetrahedron: Asymmetry 1993, 4, 2451.
31. Carlsen, P. H. J.; Aase, K.; Acta Chemica Scandinavica 1993, 47, 737.
32. Marzi, M.; Minetti, P.; Moretti, G.; Tinti, M. O.; Angelis, F.; J. Org. Chem. 2000, 65, 6766; Marzi, M.; Tinti, M. O.; de Angelis, F.; US pat. 6,420,599 B2 2002.
33. Boons, G. J.; Entwiatle, D. A;. Ley, S. V.; Woods, M.; Tetrahedron Lett. 1993, 34, 5649.
34. Jurczak, J.; Pikul, S.; Bauer, T.; Tetrahedron 1986, 42, 447.
35. Terakado, D.; Koutaka, H.; Oriyama, T.; Tetrahedron: Asymmetry 2005, 16, 1157.
36. Palomo, J. M.; Segura, R. L.; Mateo, C.; Terreni, M.; Guisan, J. M.; Fernández-Lafuente, R.; Tetrahedron: Asymmetry 2005, 16, 869.
37. Chong, J. M.; Sokoll, K. K.; Tetrahedron Lett. 1992, 33, 879.
38. Lewis, C. A.; Sculimbrene, B. R.; Xu, Y.; Miller, S. J.; Org. Lett. 2005, 7, 3021.
39. Batovska, D. I.; Tsubota, S.; Kato, Y.; Asanoa, Y.; Ubukatab, M.; Tetrahedron: Asymmetry 2004, 15, 3551.
40. Britgoff, D.; Laumen, K.; Schneider M. P.; J. Chem. Soc., Chem. Commun. 1986, 1583.
41. Ader, U.; Andersch, P.; Berger, M.; Goergens, U.; Seemayer, R.; Schneider, M.; Pure Appl. Chem. 1992, 64, 1170.
42. Murata, M.; Terao, Y.; Achiwa, K.; Nishio, T.; Seto, K.; Chem. Pharm. Bull. 1989, 37, 2670.
43. Kawanami, Y.; Honnma, A.; Ohta, K.; Matsumoto, N.; Tetrahedron 2005, 61, 693.
44. Kato, Y.; Fujiwara, L.; Asano, Y.; Bioorg. Med. Chem. Lett. 1999, 9, 3207.
45. Wong, C. H.; Matos, J. R.; J. Org. Chem. 1985, 50, 1992.
46. Schoevaart, R.; Rantwijk, F.; Sheldon, R. A.; J. Org. Chem. 2000, 65, 6940.
47. Franke, D.; Machajewski, T.; Hsu, C. C.; Wong, C. H.; J. Org. Chem. 2003, 68, 6828.
48. Terao, Y.; Tsuji, K.; Murata, M.; Achiwa, K.; Nishio, T.; Watanabe, N.; Seto, K.; Chem. Pharm. Bull. 1989, 37, 1653.
49. Partali, V.; Melbye, A. G.; Alvik, T.; Anthonsen, T.; Tetrahedron: Asymmetry 1992, 1, 65.
50. Chen, C. S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J.; J. Am. Chem. Soc. 1982, 104, 7294.
51. Vänttinen, E.; Kanerva, L. T.; Tetrahedron: Asymmetry 1997, 8, 923.
52. Sakai, T.; Kishimoto, T.; Tanaka, Y.; Ema, T.; Utaka, M.; Tetrahedron Lett. 1998, 39, 7881.
53. Kim, S. C.; Kim, Y. H.; Lee, H.; Yoon, D. Y.; Song, B. K.; J. Mol. Catal. B: Enzym. 2009, 49, 75.
54. Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C.; J. Mol. Catal. A: Chem. 2006, 257, 149.
55. Vieville, C.; Yoo, J. W.; Pelet, S.; Mouloungui, Z.; Catal. Lett. 1998, 56, 245.
56. Pallavicini, M.; Valoti, E.; Villa, L.; Piccolo, O.; J. Org. Chem. 1994, 59, 1751.
57. Lemaire, M.; Bolte, J.; Tetrahedron: Asymmetry 1999, 10, 4755.
58. Chênevert, R.; Gagnon, R.; J. Org. Chem. 1993, 58, 1054.
Recebido em 18/4/10; aceito em 21/8/10; publicado na web em 8/12/10
-
1Zhou, C. H.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q.; Chem. Soc. Rev. 2008, 37, 527.
-
2King, A. G.; Wright, M. W.; J. Chem. Educ. 2007, 84, 202.
-
3Pinto, A. C.; Guarieiro, L. N.; Rezende, M. J.; Ribeiro, N. M.; Torres, E. A.; J. Braz. Chem. Soc. 2005, 16, 1313.
-
4Santos, A. P.; Pinto, A. C.; Quimica Nova na Escola 2009, nº 31, 58.
-
5http://www.biodiesel.gov.br/, acessada em Junho 2009.
» link -
6Geris, R.; Santos, N. A. C.; Amaral, B. A.; Maia, I. S.; Castro, V. D.; Carvalho, J. R. M.; Quim. Nova 2007, 30, 1369.
-
7Mota, C. J. A.; Tchê Química 2006, 3, 26.
-
8Mota, C. J. A.; da Silva, C. X. A.; Gonçalves, V. L. C.; Quim. Nova 2009, 32, 639.
-
9http://www1.eere.energy.gov/biomass/pdfs/35523.pdf, acessada em Janeiro 2009.
» link -
10Pagliaro, M.; Rossi, M.; The Future of Glycerol - New Usages for a Versatile Raw Material, RSC Publishing: Cambridge, 2008.
-
11Katha, S. S.; Medical Hypotheses 1999, 53, 338.
-
12Lide, D. R., editor-in-chief; Handbook of Chemistry and Physics, 87th ed., Taylor and Francis Group: Boca Raton, 2006.
-
13Callam, C. S.; Singer, S. J.; Lowary, T. L.; Hadad, C. M.; J. Am. Chem. Soc. 2001, 123, 11743.
-
14Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Pina, C. D.; Angew. Chem., Int Ed 2007, 46, 4434.
-
15Wang, Z. X.; Zhuge, J.; Fang, H.; Prior, B. A.; Biotechnology Advances 2001, 19, 201.
-
16de Arruda, P. V.; Rodrigues, R. C. L. B.; Felipe, M. G. A.; Revista Analytica 2006, 26, 56.
-
17Andrews, M. A.; Klaeren, S. A.; US pat. 5,026,927 1991
-
18http://www.aboissa.com.br/, acessada em Novembro 2008.
» link -
19Araújo, Y. J. K.; de Lima, D. P.; Beatriz, A.; Orbital - The Electronic Journal of Chemistry 2009, 1, 1.
-
20Faber, K.; Biotransformation in Organic Chemistry, 4th ed., Springer: Berlin, 2000.
-
21Fessner, W-D.; Anthonsen, T., eds.; Modern Biocatalysis - Stereoselective and Environmentally Friendly Reactions, Wiley-VCH: Weinheim, 2009.
-
22Strübing, D.; Krumlinde, P.; Piera, J.; Bäckvall, J-E.; Adv. Synth. Catal. 2007, 349, 1577.
-
23Uang, B. J.; Po, S. Y.; Hung, S. C.; Liu, H. H.; Hsu, C. Y.; Lin, Y. S.; Chang, J. W.; Pure Appl. Chem. 1997, 69, 615.
-
24Debost, J. L.; Gelas, J.; Horton, D.; J. Org. Chem 1983, 48, 1381.
-
25de Oliveira, P. S. M.; Ferreira, V. F.; de Souza, M. V. N.; Quim. Nova 2009, 32, 441.
-
26Jung, M. E.; Shaw, T. J.; J. Am. Chem. Soc 1980, 102, 6304.
-
27Barreiro, E. J.; Ferreira, V. F.; Costa, P. R. R.; Quim. Nova 1997, 20, 647.
-
28Mukaiyama, T.; Tanabe, Y.; Shimizu, M.; Chem. Lett. 1984, 401.
-
29Hsu, C. Y.; Lin, Y. S.; Uang, B. J.; Tetrahedron: Asymmetry 1990, 1, 219.
-
30Buser, H. P.; Spindler, F.; Tetrahedron: Asymmetry 1993, 4, 2451.
-
31Carlsen, P. H. J.; Aase, K.; Acta Chemica Scandinavica 1993, 47, 737.
-
32Marzi, M.; Minetti, P.; Moretti, G.; Tinti, M. O.; Angelis, F.; J. Org. Chem. 2000, 65, 6766;
- Marzi, M.; Tinti, M. O.; de Angelis, F.; US pat. 6,420,599 B2 2002
- 33. Boons, G. J.; Entwiatle, D. A;. Ley, S. V.; Woods, M.; Tetrahedron Lett 1993, 34, 5649.
- 34. Jurczak, J.; Pikul, S.; Bauer, T.; Tetrahedron 1986, 42, 447.
- 35. Terakado, D.; Koutaka, H.; Oriyama, T.; Tetrahedron: Asymmetry 2005, 16, 1157.
- 36. Palomo, J. M.; Segura, R. L.; Mateo, C.; Terreni, M.; Guisan, J. M.; Fernández-Lafuente, R.; Tetrahedron: Asymmetry 2005, 16, 869.
- 37. Chong, J. M.; Sokoll, K. K.; Tetrahedron Lett 1992, 33, 879.
- 38. Lewis, C. A.; Sculimbrene, B. R.; Xu, Y.; Miller, S. J.; Org. Lett 2005, 7, 3021.
- 39. Batovska, D. I.; Tsubota, S.; Kato, Y.; Asanoa, Y.; Ubukatab, M.; Tetrahedron: Asymmetry 2004, 15, 3551.
- 40. Britgoff, D.; Laumen, K.; Schneider M. P.; J. Chem. Soc., Chem. Commun 1986, 1583.
- 41. Ader, U.; Andersch, P.; Berger, M.; Goergens, U.; Seemayer, R.; Schneider, M.; Pure Appl. Chem. 1992, 64, 1170.
- 42. Murata, M.; Terao, Y.; Achiwa, K.; Nishio, T.; Seto, K.; Chem. Pharm. Bull. 1989, 37, 2670.
- 43. Kawanami, Y.; Honnma, A.; Ohta, K.; Matsumoto, N.; Tetrahedron 2005, 61, 693.
- 44. Kato, Y.; Fujiwara, L.; Asano, Y.; Bioorg. Med. Chem. Lett 1999, 9, 3207.
- 45. Wong, C. H.; Matos, J. R.; J. Org. Chem. 1985, 50, 1992.
- 46. Schoevaart, R.; Rantwijk, F.; Sheldon, R. A.; J. Org. Chem. 2000, 65, 6940.
- 47. Franke, D.; Machajewski, T.; Hsu, C. C.; Wong, C. H.; J. Org. Chem. 2003, 68, 6828.
- 48. Terao, Y.; Tsuji, K.; Murata, M.; Achiwa, K.; Nishio, T.; Watanabe, N.; Seto, K.; Chem. Pharm. Bull. 1989, 37, 1653.
- 49. Partali, V.; Melbye, A. G.; Alvik, T.; Anthonsen, T.; Tetrahedron: Asymmetry 1992, 1, 65.
- 50. Chen, C. S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J.; J. Am. Chem. Soc. 1982, 104, 7294.
- 51. Vänttinen, E.; Kanerva, L. T.; Tetrahedron: Asymmetry 1997, 8, 923.
- 52. Sakai, T.; Kishimoto, T.; Tanaka, Y.; Ema, T.; Utaka, M.; Tetrahedron Lett. 1998, 39, 7881.
- 53. Kim, S. C.; Kim, Y. H.; Lee, H.; Yoon, D. Y.; Song, B. K.; J. Mol. Catal. B: Enzym. 2009, 49, 75.
- 54. Aresta, M.; Dibenedetto, A.; Nocito, F.; Pastore, C.; J. Mol. Catal. A: Chem 2006, 257, 149.
- 55. Vieville, C.; Yoo, J. W.; Pelet, S.; Mouloungui, Z.; Catal. Lett 1998, 56, 245.
- 56. Pallavicini, M.; Valoti, E.; Villa, L.; Piccolo, O.; J. Org. Chem. 1994, 59, 1751.
- 57. Lemaire, M.; Bolte, J.; Tetrahedron: Asymmetry 1999, 10, 4755.
- 58. Chênevert, R.; Gagnon, R.; J. Org. Chem. 1993, 58, 1054.
Datas de Publicação
-
Publicação nesta coleção
02 Mar 2011 -
Data do Fascículo
2011
Histórico
-
Recebido
18 Abr 2010 -
Aceito
21 Ago 2010