Abstracts
Interactions of some diimine copper(II) complexes with bovine serum albumin (BSA) were investigated by spectroscopic techniques in order to compare the stability of the complexes and their capability of causing oxidative damage to the protein. The tri- and tetradentate imine ligands employed in this work contain pyridine, pyrazine or imidazole moieties, which are ubiquitous in biological systems. The relative thermodynamic stabilities of the copper(II) complexes were estimated by circular dichroism (CD) using BSA as the competitive ligand. The apparent stability constants determined for the complexes are very similar to one another and to that of the Cu(BSA) complex itself, for which log K Cu(BSA) = 12.9 has already been described in the literature, indicating that the complexes are quite stable under physiological conditions. Two different copper binding sites were evidenced on BSA by spectroscopic measurements (CD, UV-Vis and EPR), depending on the ligand and on the [CuL]:[protein] stoichiometric ratio. Metal binding to any of the sites gives rise to significant protein oxidative damage, especially in the presence of hydrogen peroxide, indicating an oxidative process based on reactive oxygen species (ROS). A small amidated peptide, Asp-Thr-His-NH2, corresponding to the N-terminal region of BSA was synthesized, and its interaction with all the diimine-copper(II) complexes was also investigated in order to clarify the copper imine complex-albumin interactions. Electronic structure calculations at the density functional theory (DFT) level were made to compare the copper-ligand binding energies for each complex with that of the metal coordinated at the N-terminal site of the protein.
copper(II) complexes; BSA; spectroscopy; binding energies; oxidative damage; DFT
Interações de alguns compostos di-imínicos de cobre(II) com albumina de soro bovino (BSA) foram investigadas por técnicas espectroscópicas, visando comparar a estabilidade dos complexos e verificar a sua capacidade de causar danos oxidativos à proteína. Os ligantes imínicos tri- e tetradentados utilizados neste trabalho contêm grupamentos piridina, pirazina ou imidazol, frequentemente encontrados no meio biológico. A estabilidade termodinâmica relativa de cada um dos complexos foi determinada através de dicroísmo circular (CD), usando a própria BSA como ligante competitivo. Os valores estimados das constantes de equilíbrio aparentes foram muito semelhantes entre si e ao da espécie Cu(BSA), para a qual log K Cu(BSA) = 12.9 já foi relatado na literatura, mostrando que os complexos estudados são bastante estáveis em condições fisiológicas. Dois diferentes sítios de coordenação do íon cobre foram verificados nestes estudos, evidenciados por medidas espectroscópicas (UV-Vis, CD e EPR), dependendo do ligante e da proporção estequiométrica [CuL]/[BSA] utilizada. Entretanto, a inserção do íon cobre em qualquer dos sítios ligantes ocasionou danos oxidativos significativos à proteína, especialmente em presença de peróxido de hidrogênio, indicando um processo oxidativo com formação de espécies reativas de oxigênio (EROs). Um tripeptídeo amidado, Asp-Thr-His-NH2, mimético do sítio N-terminal da BSA, foi sintetizado e a investigação de sua interação com os complexos de cobre permitiu uma compreensão melhor do sistema cobre-BSA. Cálculos de estrutura eletrônica empregando a teoria do funcional de densidade (DFT) foram efetuados para determinar as energias de ligação do cobre em cada complexo e no sítio N-terminal da proteína, buscando correlacioná-las com os dados de estabilidade termodinâmica.
ARTICLE
Interactions of di-imine copper(II) complexes with albumin: competitive equilibria, promoted oxidative damage and DFT studies
M. Amélia A. AzzelliniI; Mariana P. AbbottI; Alessandra Machado* * Present Address: Universidade Federal Tecnológica do Paraná, 85601-970 Francisco Beltrão-PR, Brasil ,II; M. Terêsa M. MirandaII; Leone C. GarciaIII; Giovanni F. CaramoriI,III; Marcos B. GonçalvesIV; Helena M. PetrilliIV; Ana M. C. Ferreira** ** e-mail: amdcferr@iq.usp.br ,I
IDepartamento de Química Fundamental, Universidade de São Paulo, 05508-000 São Paulo-SP, Brazil
IIDepartamento de Bioquímica, Instituto de Química, Universidade de São Paulo, 05508-000 São Paulo-SP, Brazil
IIIDepartamento de Química, Universidade Federal de Santa Catarina, 88040-900 Florianópolis-SC, Brazil
IVInstituto de Física, Universidade de São Paulo, 05314-970 São Paulo-SP, Brazil
ABSTRACT
Interactions of some diimine copper(II) complexes with bovine serum albumin (BSA) were investigated by spectroscopic techniques in order to compare the stability of the complexes and their capability of causing oxidative damage to the protein. The tri- and tetradentate imine ligands employed in this work contain pyridine, pyrazine or imidazole moieties, which are ubiquitous in biological systems. The relative thermodynamic stabilities of the copper(II) complexes were estimated by circular dichroism (CD) using BSA as the competitive ligand. The apparent stability constants determined for the complexes are very similar to one another and to that of the Cu(BSA) complex itself, for which log KCu(BSA) = 12.9 has already been described in the literature, indicating that the complexes are quite stable under physiological conditions. Two different copper binding sites were evidenced on BSA by spectroscopic measurements (CD, UV-Vis and EPR), depending on the ligand and on the [CuL]:[protein] stoichiometric ratio. Metal binding to any of the sites gives rise to significant protein oxidative damage, especially in the presence of hydrogen peroxide, indicating an oxidative process based on reactive oxygen species (ROS). A small amidated peptide, Asp-Thr-His-NH2, corresponding to the N-terminal region of BSA was synthesized, and its interaction with all the diimine-copper(II) complexes was also investigated in order to clarify the copper imine complex-albumin interactions. Electronic structure calculations at the density functional theory (DFT) level were made to compare the copper-ligand binding energies for each complex with that of the metal coordinated at the N-terminal site of the protein.
Keywords: copper(II) complexes, BSA, spectroscopy, binding energies, oxidative damage, DFT
RESUMO
Interações de alguns compostos di-imínicos de cobre(II) com albumina de soro bovino (BSA) foram investigadas por técnicas espectroscópicas, visando comparar a estabilidade dos complexos e verificar a sua capacidade de causar danos oxidativos à proteína. Os ligantes imínicos tri- e tetradentados utilizados neste trabalho contêm grupamentos piridina, pirazina ou imidazol, frequentemente encontrados no meio biológico. A estabilidade termodinâmica relativa de cada um dos complexos foi determinada através de dicroísmo circular (CD), usando a própria BSA como ligante competitivo. Os valores estimados das constantes de equilíbrio aparentes foram muito semelhantes entre si e ao da espécie Cu(BSA), para a qual log KCu(BSA) = 12.9 já foi relatado na literatura, mostrando que os complexos estudados são bastante estáveis em condições fisiológicas. Dois diferentes sítios de coordenação do íon cobre foram verificados nestes estudos, evidenciados por medidas espectroscópicas (UV-Vis, CD e EPR), dependendo do ligante e da proporção estequiométrica [CuL]/[BSA] utilizada. Entretanto, a inserção do íon cobre em qualquer dos sítios ligantes ocasionou danos oxidativos significativos à proteína, especialmente em presença de peróxido de hidrogênio, indicando um processo oxidativo com formação de espécies reativas de oxigênio (EROs). Um tripeptídeo amidado, Asp-Thr-His-NH2, mimético do sítio N-terminal da BSA, foi sintetizado e a investigação de sua interação com os complexos de cobre permitiu uma compreensão melhor do sistema cobre-BSA. Cálculos de estrutura eletrônica empregando a teoria do funcional de densidade (DFT) foram efetuados para determinar as energias de ligação do cobre em cada complexo e no sítio N-terminal da proteína, buscando correlacioná-las com os dados de estabilidade termodinâmica.
Introduction
Albumin, a plentiful protein (66 kDa) in mammalian blood serum, is involved in the transport of different substrates, including hormones, xenobiotics, drugs and metals.1 Human and bovine albumins (HSA and BSA, respectively) contain the N-terminal sequence X-Y-His that constitutes a strong physiological binding site for endogenous metal ions, and has been extensively investigated.2-5 Particularly, the equilibrium constant for the binding of copper(II) ions to this high affinity site of BSA, which involves both histidyl and carboxyl groups, has been estimated by different groups6,7 as log KCu(BSA) = 11.12 or 12.9. Analogously, a very recent work8 estimated the affinity of the N-terminal site in HSA for copper as 1 pmol L-1 or as log KCu(HSA) = 12.0.
Earlier studies have also indicated further non-specific copper(II) binding sites in HSA and BSA,5 while more recent investigations showed that endogenous zinc(II) ions and toxic nickel(II) and cadmium(II) ions can displace copper from a second specific metal site, but not from the primary N-terminal site.9 This second binding site in albumin was described as multi-metal, relevant as zinc(II) binding site, and a target for toxic cations such as nickel(II) and cadmium(II). On the other hand, studies based on isothermal titration calorimetry showed that at higher pH (ca. 9) a second copper(II) ion binds to the protein in a tetragonal site, through a thiolate ligation, probably to deprotonated Cys-34.8 However, in all these previous studies the copper ions were added to the protein as the corresponding aqua-complex (as chloride or sulphate salt),4-9 and the influence of different ligands in very stable copper complexes has not been sufficiently investigated yet.
A few studies have been reported about the interactions of copper complexes with albumin. Based on absorption titration data, the binding constant of an imine-copper(II) complex with BSA has been reported as (2.4 ± 0.3)×103 mol-1 L.10 In the presence of oxygen, the site-specific cleavage of the protein resulted in fragments of 49, 45, 22 and 17 kD, indicating the presence of two specific binding sites for the copper complex. More recently, the data collected in a study of a copper(II) complex with a compartmental Schiff base ligand indicated that the reduced form CuIL is bound ca. 4 times more strongly to BSA than the corresponding oxidized form CuIIL.11 Particularly, the copper complex [CuI(o-phen)2]+, or [CuII(o-phen)2]2+ in the presence of β-mercaptoethanol, o-phen = 1,10-phenanthroline, has been described to promote protein degradation.12 Indeed, peptides and amino acids were produced when α-casein and BSA were incubated in the presence of such complexes. Further, the replacement of o-phen by ethylenediaminetetraacetate ion (edta), 2,2-bipyridine, or 2,9-dimethyl-1,10-phen, or substitution of copper(II) by iron(III), cobalt(II) or nickel(II) hampers protein degradation.
Since copper is a redox active essential metal that can be reduced by some protein groups and subsequently re-oxidized by molecular oxygen, some oxidative damage to proteins can arise from their binding to this metal ion. BSA is known as a very weak reductant of copper(II), producing very low levels of peroxide. However, Cu+ ions bound to proteins can act as reductant. Additionally, if coordinated to appropriate ligands, such as imines that stabilize both oxidation states usually found in biological medium, the process could be modulated by the type of environment provided by the coordinating framework to the copper ion.13
All the factors predominant in managing the reactivity of copper centers inserted in proteins (electronic configurations, redox potentials, steric characteristics, etc.) became particularly relevant in recent years. Proteins such as copper, zinc superoxide dismutase (SOD1) in its mutated varieties showed additional metal binding sites and enhanced peroxidase activity when those sites were metallated. Further, β-amyloid protein also exhibited good coordinating properties, especially for copper, iron and zinc ions. Consequently, a potential relationship with severe neurodegenerative diseases was reported for both classes of proteins.14-16 Therefore, designed ligands with high affinity for copper ions and competitive coordinating metal capacity compared to proteins and other biomolecules can be of great interest for diseases in which metal ions facilitate undesirable oxidation and aggregation of proteins or related oligopeptides.
Recently, we studied a series of oxindolimine-copper(II) complexes comparing their ability to interact with human serum albumin (HSA), causing changes in its secondary structure, and consequently promote significant oxidative damage. These results indicated that the more stable complexes, when compared to the N-terminal metal binding site, are also those more reactive on oxidizing the protein.17
Herein, the interactions of a series of diimine copper(II) complexes with BSA were investigated using different spectroscopic techniques, UV-Vis, CD and EPR, with the aim of verifying the influence of this type of ligand in the prevalent metal insertion site and the extension of consequent protein oxidative damage. These complexes have already shown antioxidant as well as pro-oxidant properties, acting as efficient mimics of SOD1, but additionally generating reactive oxygen species (ROS) in significant extension.18,19 This type of reactivity in copper(II) complexes is particularly relevant not only for the study of its possible implication in neurodegenerative diseases,20 but also for the development of potential metallopharmaceuticals with ability of generating or scavenging reactive species by interaction with specific biological targets.21 Complementing these data, a small amidated peptide corresponding to the N-terminal region of BSA was synthesized, and its interaction with all these diimine-copper(II) complexes was investigated in order to clarify the copper imine complex-albumin interactions. Further, DFT calculations were made to estimate the CuII-L binding energies for each ligand, in order to explain their relative stability regarding that of copper bound to the N-terminal metal binding site in the protein.
Materials and Methods
Materials
The reagents 2-acetylpyridine, acetylpyrazine, diacetyl, histamine, 2-(2-aminoethyl)pyridine, copper(II) perchlorate hexahydrate, 5,5'-dimethyl-3,4-dihydropyrrole-N-oxide (DMPO) and bovine serum albumin (BSA) were purchased from Aldrich Chem. Co. Inc. (Milwaukee, WI, USA). Sulfhydryl groups in BSA were quantified using 5,5-dithiobis-(2-nitrobenzoic acid), with ε (at 412 nm) = 13.6 L mmol-1 cm-1.22 A typical sulfhydryl to BSA molar ratio (R) of 0.6-0.7 was obtained, in accordance with values verified in fresh plasma albumin.23 The spin scavenger DMPO was previously purified by a reported method.24 SOD1 (from bovine erythrocytes), diisopropylethylamine (dipea) and 2,4-dinitrophenylhydrazine (DNPH) were from Sigma Chem. Co. (St. Louis, MI, USA). Inorganic phosphates, used in the buffer solutions, and histidine were from Merck (Darmstadt, GE), and hydrogen peroxide 35% in weight, free of stabilizers, was obtained from Peróxidos do Brasil Ltd. (São Paulo, Brazil). Solutions of this reagent, prepared by appropriate dilution of the commercial product, were analyzed by the metavanadate method.25 Benzotriazole-1-yl-oxy-tris(dimethylamino)phosphonium hexafluorophosphate (bop) and the amino acid derivatives were bought from Bachem (Torrance, CA, USA). The 4-methylbenzhydrylamine resin (MBHA) was from Applied Biosystems (Foster City, CA, USA). Most of the organic solvents were of analytical grade and purchased from Merck (Darmstadt, Germany). Acetonitrile and trifluoroacetic acid (tfa) were of chromatographic grade, and also from Merck. Deionized water from a Barnstead D 4700 apparatus or from a Milli-Q deionizer was used to prepare the solutions.
Syntheses and characterization of the metal complexes
The diimine or Schiff base copper(II) compounds used in these studies (see structures in Scheme 1) were prepared by standard methods already described,26 with suitable modifications. For the tridentate imine ligands, small changes were introduced, especially the strict control of the pH of reaction. These complexes have been characterized by different spectroscopic techniques, including UV-Vis, IR and EPR, besides elemental analysis and molar conductivity measurements, as described elsewhere.18,19,27
Peptide synthesis, purification and chemical characterization
The tripeptide Asp-Thr-His-NH2 was synthesized manually by the stepwise solid-phase method using the Boc (or tert-butyloxycarbonyl) chemistry and customized protocols.28,29 The side-chain protecting groups were: cyclohexyl for aspartic acid, benzyl for threonine and benzyloxymethyl for histidine. Briefly, the peptide chain was built-up on the MBHA resin at room temperature employing 2.5-fold excess of the incorporating amino acids, equimolar amounts of bop and volumes of dipea sufficient to maintain the pH of the reaction media around 9.0.30t-Boc removal was carried-out for 20 min in 50% tfa/dichloromethane (dcm) containing 1% anisol, while neutralization of the free amine group was achieved in 10% triethylamine (tea)/dcm. The steps of coupling, recoupling, partial deprotection and neutralization were monitored by the ninhydrin test.31 Final cleavage from the resin and simultaneous full deprotection was conducted at 0 ºC for 90 min, in HF containing 1% anisol.29
Analysis and purification of the crude peptide were carried out using the detection wavelength of 220 nm, flow rates and linear gradients of solvents A and B (where A was 0.1% tfa/water and B was 60% tfa/water containing 0.09% tfa). The fractions that presented equivalent peptide purity were pooled and lyophilized. The chemical identity of the resulting purified peptide was confirmed by electrospray ionization mass spectrometry (ESI-MS) and amino acid analysis. The [M+1H]+ peak obtained was identical to the expected value of 371.0. The results of amino acid analysis, with the expected values given in parentheses, were: Asp, 1.1(1.0); Thr, 0.8(1.0); His, 1.0(1.0).
Methods and instruments
Electronic spectra were registered in a Shimadzu 1650PC UV/Vis spectrophotometer, using 10 mm length cuvettes. EPR spectra were obtained in an EMX spectrometer from Bruker Instruments, operating at X-band (9.33 GHz), modulation frequency 100 kHz and power of 20.17 mW. For these measurements, the isolated copper complexes were dissolved in dimethylformamide (dmf), acetonitrile or methanol:water mixtures (3:1, v/v), and the solution frozen at 77 K, in Wilmad suprasil tubes. Modulation amplitude of 15G was usually employed. For the experiments in the presence of BSA, spectra were registered immediately after addition of the copper complex (dissolved in appropriate solvent) to the protein dissolved in buffer solution (0.70 mmol L-1), in stoichiometric ratio [CuL]:[BSA] up to 2:1, and after incubation for 30 min at room temperature. The magnetic field was calibrated with 2,2-bis(4-tert-octylphenyl)-1-picrylhydrazyl (DPPH) as external standard (g = 2.0036). In the detection of hydroxyl radicals by the spin-trapping method, 5,5-dimethyl-pirroline-N-oxide (DMPO) was used as spin scavenger.32 Spectra were registered with modulation amplitude of 1 G, using standard flat cells at room temperature, after 10 min of reaction with hydrogen peroxide at (25.0 ± 0.5) ºC. Concentrations of the [DMPO-OH] adduct were estimated by double integration, using 4-hydroxy-2,2,6,6-tetramethyl-1-piperidinyloxy (tempol, 38.2 µmol L-1) for calibration. CD spectra were recorded on a JASCO J-720 or a Jobin Yvon CD6 spectropolarimeter in the range 300-650 nm, using spherical cells with 10 mm of optical length. BSA solutions (0.30 to 0.60 mmol L-1 in phosphate buffer 50 mmol L-1, pH 7.4) were employed in the presence of variable concentrations (0 to 0.60 mmol L-1) of the copper complexes. Perchlorate copper(II) salt, added as the aqua complex, was used in control experiments. Reversed-phase HPLC (RP-HPLC) was carried out in two different equipments: a LDC analytical system coupled to a Vydac C4 column (5 µm, 300Å, 0.46 id × 25.00 cm) and a semi-preparative Beckman System Gold coupled to a Vydac C4 column (5 µm, 300 Å, 2.2 id × 25.00 cm), both from The Separation Group, Inc. (Hesperia, CA, USA).
Carbonyl group assay
Formation of carbonyl groups from the oxidative damage to amino acids was detected by the corresponding dinitrophenylhydrazones, after reaction with DNPH.33 Samples were prepared by adding 300 µL of BSA (1.00 mmol L-1), 200 µL of the copper complex solution (3.0 mmol L-1) and 200 µL of hydrogen peroxide (30 mmol L-1) into 300 µL of phosphate buffer (50 mmol L-1, pH 7.35). For the experiments performed in the absence of hydrogen peroxide, 500 µL of phosphate buffer were used, and in the control experiments only hydrogen peroxide was added to the BSA in buffer solution. After incubation for 30 min at (37.0 ± 0.5) ºC, 1.00 mL of DNPH solution (1 mmol L-1 in HCl) was added to each 1.00 mL of sample. The final solutions were incubated for more 30 min at (37.0 ± 0.5) ºC, followed by addition of 1.50 mL of 1.0 mol L-1 NaOH solution. The absorbance at 450 nm (ε = 2.2×104 L mol-1 cm-1) was then measured after 5 min, according to the procedure described by Hawkins and Davies.34 A similar procedure was used in the case of the tripeptide Asp-Thr-His-NH2 (or DTH): 300 µL of buffer solution (50 mmol L-1, pH 7.35), 300 µL of the peptide (3.00 mmol L-1), 200 µL of the copper(II) complex (2.0 mmol L-1) and 200 µL of hydrogen peroxide (20 mmol L-1) were mixed in a final volume of 1.00 mL.
SOD activity measurements
The inhibition of nitroblue tetrazolium (NBT2+) reduction by superoxide radicals was used to monitor the SOD activity of the copper(II) complexes studied.35,36 Superoxide radicals were generated by the system xanthine/xanthine oxidase, and the formazan formation was measured at 530 nm in phosphate buffer (pH 7.4), in the absence and in the presence of different amounts of the copper complexes. One nitroblue tetrazolium unit of SOD activity corresponds to the amount of enzyme that leads to 50% inhibition of NBT2+ reduction (IC50).37 Similar results with these copper(II) complexes have been obtained in a previous work.19
Computational methods
Binding energies were calculated using the projector augmented wave (PAW) method as embodied in the CP-PAW code.38 We have previously shown for similar Cu complexes17 that binding energies obtained using the CP-PAW code (PBE/PAW) and Gaussin03 package39 (BP86/TZVP) agree with less than 7% difference. Since the procedure used is the same as described in a previous work,17 only the binding energies obtained with the CP-PAW and the topological analysis of the electron density with Gaussian03 are shown here. The ab-initio PAW is an all-electron method in the Kohn-Sham scheme of DFT, where the wave function is expanded into an augmented plane wave basis set. To simulate the systems as truly isolated molecules, the repeated supercell approach was used. To prevent wave function overlap and artificial electrostatic interactions between periodic images, a minimum distance of 6 Å was kept between periodic images, and the Blöchls charge decoupling scheme was used.40 Geometry optimizations were performed using 40Ry and 160Ry as the "cut off" energies in the corresponding expansions of the wave functions and charge densities, with the generalized gradient approximation (GGA) of Perdew and Burke-Ernzerhof (PBE).41 It was used one s projector function per angular momentum for the hydrogen atom; for carbon, nitrogen and oxygen atoms two s, two p, and one d were considered; and an additional d-projector function was used for copper. For all copper(II) complexes, the geometry optimizations were carried out without symmetry constraints.
The metal-ligand bonding of complexes was investigated in the light of the quantum theory of atoms in molecules, QTAIM,42 employing the gradient-correct BP86 functional43 with all-electron GTO TZVP basis set.44 The QTAIM analysis was carried out with the AIMPAC suite of programs.45
The charge distribution in complexes 1-6 and [Cu(BSA)]2+ was analysed with the QTAIM method, which explores the electron density topology in molecular systems, providing insightful information on the nature of chemical bonds. According to the QTAIM, the definition of the chemical bond is based on the existence of a bond critical point (BCP) along a line of maximum density (bond path) between two atomic centres. At the BCP the gradient of the density vanishes, ∇ρb = 0, and the sum of the eigenvalues of the density Hessian matrix yields the Laplacian, ∇2ρb. The topology of the ∇2ρb characterizes local concentrations (∇2ρb < 0) of the electron density and its depletion (∇2ρb > 0). In interactions where ∇2ρb < 0 (shared interactions), a lowering of the potential energy density Vb associated with a concentration in charge between the nuclei along the bond path is observed. On the other hand, closed-shell interactions, ∇2ρb > 0, are dominated by the kinetic energy density Gb in the region of the interatomic surface. Additional information about chemical bond type is available from the total electronic energy density Hb = Gb +Vb at the BCP. According to Bianchi et al.46 the topological results permit us to classify the atomic interactions as following: i) shared interactions (ρb high and ∇2ρb < 0) which can be reclassified as covalent bonds (Vb << 0, Gb << |Vb|, Hb << 0) and polar-covalent bonds (Vb << 0, Gb < |Vb|, Hb << 0); ii) closed-shell interactions (ρb low and ∇2ρb > 0), classified as dative bonds (Vb < 0, Gb≅ |Vb|, Hb < 0), metallic bonds (Vb < 0, Gb≅ |Vb|, Hb <0, |Hb| ≅ 0), ionic bonds (Vb < 0, Gb≅ |Vb|, Hb > 0 |Hb| ≅ 0), and van der Waals interactions (Vb < 0, Gb≅ |Vb|, Hb > 0 |Hb| ≅ 0). The donor-acceptor bonds have topological parameters close to those of the metallic bond, but less shifted toward the ionic interaction.
Copper(II)-L binding energies and ligand deformation energies
For each complex, the copper(II)-L binding energy (ΔEb) was calculated according to the following equation:

In this expression, ECu(II)L is the total energy of the CuIIL, while ECu(II) is the energy of isolated CuII. The last term is the summation over the energies (Elig(i)opt) of the n ligands isolated and optimized without CuII. The deformation energy (ΔEdef) gives a comprehensive characterization of the ligand distortion, and is computed as the energy difference between the ligand in the conformation of the complex (Elig(i)) and the optimized ligand (Elig(i)opt).

Results
The present study ponders different interactions that occur in vitro, involving diimine copper(II) complexes and BSA. In Figure 1, the CD spectra of BSA solution titrated with some of the complexes studied (complexes 1 and 2) are shown, with the corresponding parameters in Table 1. CD spectra for compounds 2 to 6 were very similar, and showed bands at 570 nm, 490 nm and 320 nm (Figure 1B).

Relative stability constants
In order to compare the thermodynamic stability of the studied complexes, BSA was used as a competitive ligand for the copper ions. Titration experiments of BSA (0.3 to 0.7 mmol L-1) with the different copper-diimine complex solutions, up to 0.3 mmol L-1 in phosphate buffer (pH 7.4) and at (25.0 ± 0.2) ºC, were performed and monitored by CD spectroscopy. Copper-aqua complex, [Cu(H2O)4]2+, was used in the control experiments, in order to monitor and calculate the concentration of [Cu(BSA)] species formed, since in this case a negligible concentration of the copper ions remains non-coordinated to the N-terminal site of the protein. Based on the CD spectra data collected, the relative thermodynamic stability or formation constants for the different complexes were estimated according to the following equilibria:
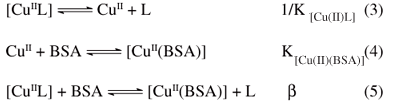
where7β = KCu(II)BSA/KCu(II)L = [Cu(BSA)] [L] / [BSA] [CuL] and KCu(II)BSA = 7.94×1012.
By using the appropriate stoichiometric relations, it was considered that:
[L] = [Cu(BSA)]
[Cu(BSA)] = [CuL]o [CuL]
[BSA] = [BSA]o [Cu(BSA)],
where [CuL]o and [BSA]o are initial or total concentrations of the diimine copper(II) complex and albumin, respectively.
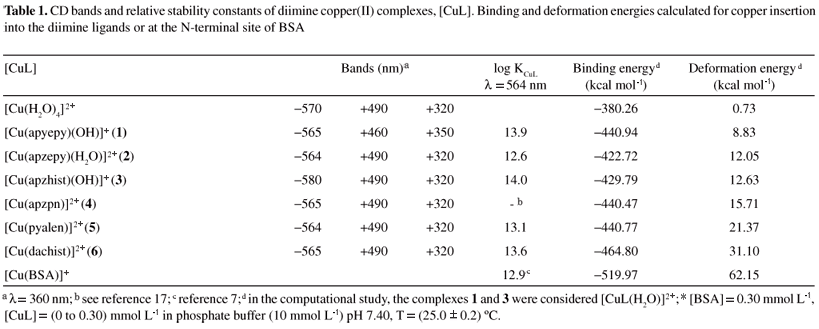
Control experiments using [Cu(H2O)4]SO4 as the added copper reagent led to the determination of a maximum [Cu(BSA)] concentration formed by plots of CD signal amplitude (at 564 nm) versus [CuL] added. A linear fit was observed: CD signal amplitude = 6.20×10-3 + 7.75 [CuL], with r = 0.995. Corresponding values of log KCuL were then obtained for each complex, and compared to the known7,9 log KCu(II)BSA = 12.9 as shown in Table 1.
Spectroscopic characterization of diimine copper(II) binding sites to BSA
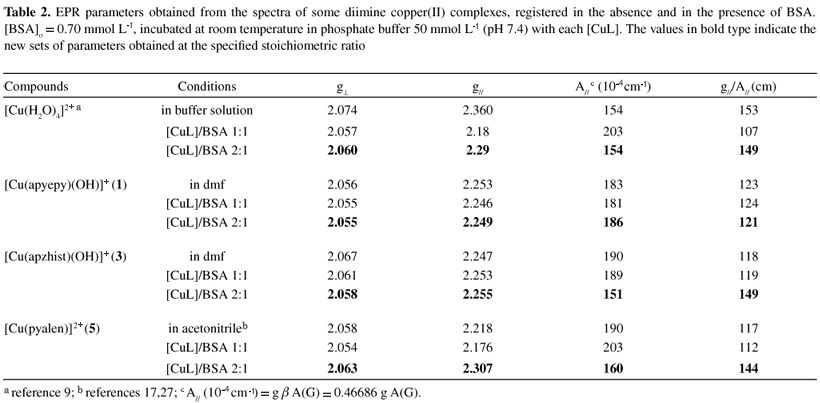
Complementing the CD data, EPR parameters for the different diimine copper(II) complexes were compared to those determined in BSA solutions (0.60 mmol L-1) after 30 min of incubation at stoichiometric amounts of [CuL]/[BSA] 1:1 and 2:1. Modifications in the spectra were observed only in the first 20 min, although monitored for 2 h. Initially, a new species attributed to the copper interaction at the N-terminal site of the protein was detected. With increasing amounts of copper ions added up to ratio 2:1, a second species with different parameters was also detected. With higher stoichiometric ratios no further changes in the spectra were observed. The characteristic data found for some of the complexes, based on EPR spectra observed, are shown in Table 2. For complexes 3 and 5, the results were similar to those obtained when the aqua complex [Cu(H2O)4]2+ was used,9 indicating at least two copper ions with different environmental geometries. In addition to a set of observed parameters (g|| = 2.18-2.25 and A|| = 190-200×104 cm-1) assigned to the copper binding at the primary site of the protein, a second set was verified with g|| = 2.25-2.30 and A|| = 150-170×104 cm-1 when a secondary site was loaded. However, for complex 1 at all metal:protein ratios used (up to 3:1), the observed parameters were those characteristic of the first site (g|| = 2.25 and A|| = 190-200×104 cm-1), indicating that this complex 1 can load only the primary binding site for copper ions in BSA. The determined EPR parameters were optimized by simulation of the spectra using the WINEPR Simfonia program from Bruker Analytische Messtechnikas, as described previously for similar complexes.17
Furthermore, electronic spectra registered during the titration of BSA with the diimine copper(II) complexes showed different behavior, depending on the ligand. The complexes [Cu(apzepy)(H2O)]2+ (2) and [Cu(dachist)]2+ (6) exhibited spectral changes very similar to those observed when the aqua complex [Cu(H2O)4]2+ was added to the BSA solution. A shift of the d-d band from 700 to 556 nm was observed, with an increasing absorption at this wavelength being verified up to [CuL]/[BSA] = 1:1, while the solution turned to reddish. Further addition of the copper complex, up to ratio 3:1, led to the growth of a second absorption band around 640 nm, attributed to the load up of another metal binding site. However, in contrast to what was observed with the aqua complex, a noticeable simultaneous increase in the absorption at 320-350 nm was verified, which can be indicative of an additional interaction with the protein. The same behavior was observed for complex [Cu(apzhist)(OH)]+ (3), but in this case the solution turned blue.
On the other hand, compound [Cu(apyepy)(OH)]+ (1) showed peculiar spectral changes when increasing concentrations of the [CuL] were added to BSA, with growing bands at 600 nm and 400 nm, an isosbestic point at 350 nm, and a decreasing band at 310 nm, as shown in Figure 2. Very similar spectral changes have been observed with this complex in previous spectroelectrochemical experiments,27 allowing us to assign the band around 400 nm to the reduction of the diimine copper(II) species to the corresponding diimine copper(I), owing to interaction with the protein. In this case, the solution turned green, and the data suggested that only the primary metal binding site in the protein was loaded, even when the [CuL]/[BSA] ratio attained 3:1.
The interaction of compounds [Cu(apzpn)]2+ (4) and [Cu(pyalen)]2+ (5) with BSA has been previously verified.18 The addition of complex 4 to the protein caused a significant enhancement of the d-d band, concomitant with a yellow color intensification of the solution. Such improvement in the d-d band was larger than that expected or verified when a similar experiment was performed with the aqua complex. These results, in addition to the CD data, were then interpreted as the insertion of the whole complex species (metal and coordinated imine ligand) into the protein, ruling out the estimation of the relative stability constant by the methodology used.
For complex [Cu(pyalen)]2+ (5), the d-d band shifted from 670 to 500 nm (see Figure 3), and the solution became at first reddish, then orange, yellow and finally brown, simultaneously to the emergence of a band at 480 nm, which can also indicate a reduction of the copper centre (shown in Figure 3A). After the addition of this complex up to [CuL]/[BSA] ratio = 3:1, further spectral variations with time were observed, with decreasing of the band at 480 nm (Figure 3B). This kinetic effect was ascribed to the action of molecular oxygen, re-oxidizing the copper complex previously reduced by the interaction with the protein. To clarify this point, complex 5 was reduced by addition of glutathione in a supplementary experiment, and the subsequent decreasing in the absorbance was registered (see Figure 3C).

These data are in accordance with and complement the preceding reported results obtained in the CD and EPR experiments, indicating a quite different behavior of the focused copper complexes towards albumin, very dependent on the imine ligand.
Pro-oxidant and antioxidant properties
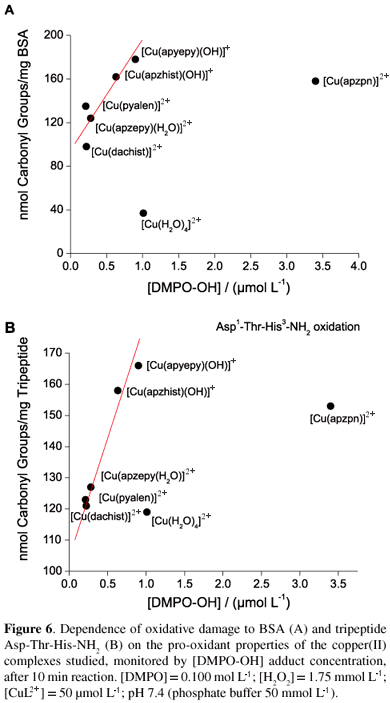
In order to better characterize the type of interactions between the diimine copper(II) complexes and BSA, the protein oxidative damage in the presence of such complexes was monitored by the formation of carbonyl groups. The corresponding data are shown in Figure 4. As can be seen, some of the complexes caused oxidative damage to the protein after incubation for 30 min at 37.0 ºC in phosphate buffer (pH 7.30), significantly more extensive than the aqua complex (species 1, 2, 3 and 5), while others exhibited similar damage (species 4 and 6). Furthermore, in the presence of hydrogen peroxide, this damage was increased in all cases.
When comparing the reactivity of these compounds towards the synthetic tripeptide Asp-Thr-His-NH2, an amidated form of fragment 1-3 of BSA, quite similar results were obtained, as shown in Figure 5. Complex [Cu(apzhist)(OH)]+ (3), which had been highly reactive towards deoxyribose19 and amino acids (data not shown), showed also high reactivity, both in the absence and in the presence of hydrogen peroxide. On the contrary, complexes [Cu(apyepy)(OH)]+ (1) and [Cu(apzpn)]2+ (4) were particularly reactive in the presence of the peroxide, but in its absence they were even less reactive than the aqua complex. Complexes 5 and 6 exhibited reactivity quite similar to that of the aqua complex.
Since different ROS can be generated during the process of oxidative injury to BSA, specially a bound copper-hydroxyl radical intermediate, [Protein-CuII(·OH)], the reactivity of the complexes studied upon hydrogen peroxide and superoxide radicals was also compared. In Figure 6, the oxidative damage observed towards BSA was plotted versus the hydroxyl generation capacity, expressed as concentration of [DMPO-OH] adduct formed in the presence of hydrogen peroxide. All of them are capable of generating hydroxyl radicals in significant amounts, and the verified oxidative damage to the protein seems to be quite dependent on this characteristic. Only two species do not fit the curve, the aqua and [Cu(apzpn)]2+ (4) complexes.
On the contrary, when the oxidative damage observed towards BSA was plotted versus the SOD activities previously determined for these copper(II) complexes (expressed as IC50),18 no significant influence of this antioxidant property was detected for all studied complexes (data not shown).
Computational studies
Since the geometry of the copper(II)-L complexes studied here were not available in the literature (neither from X-ray crystallography nor NMR studies), it has been obtained through computer simulations. The geometry of each complex was optimized using at least two initial atomic positions, and that one with the lowest total energy was chosen (however, these energy differences were smaller than 3 kcal mol-1). In order to maintain the same +2 e total charge in the Schiff base complexes series, all the complexes were considered coordinated by the imine ligand and an aqua ligand, even if some of them (complexes 1 and 3) were isolated in the hydroxo form. In all calculations the total spin was fixed in ½ ħ.
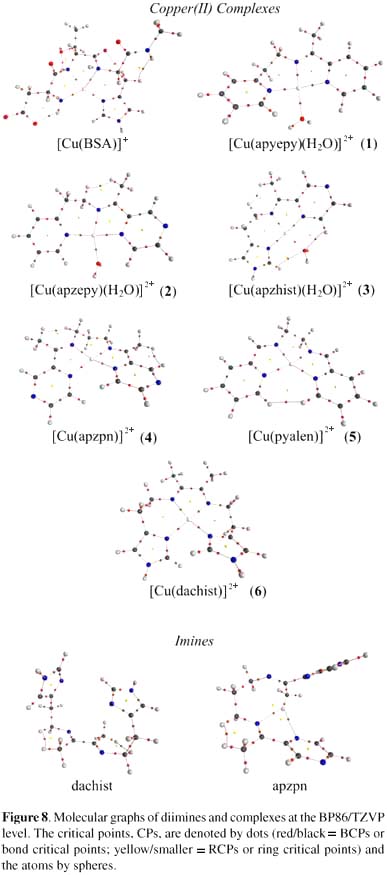
The obtained values for the binding and deformation energies in all studied copper complexes are shown in Table 1, and the respective minimum structures in Figure 7. These data pointed to a competition of the diimine ligands and the BSA N-terminal site for the copper ions, similarly to that observed in a previous work for correlated Schiff base ligands.17
The structure of the N-terminal BSA site was modelled using the three first amino acids Asp1-Thr2-His3. Due to the many degrees of freedom there are several possible structures for this N-terminal mimetic; here, the square planar coordination geometry was considered in order to better correlate with the Schiff base ligands. In this structure, metal coordination occurs by 4N in a square-planar geometry, with deprotonated amide nitrogens of Ala and His residues, in addition to the N-terminal amine and the His imidazole donor (the so-called 4N complex). The carboxyl group interacts only indirectly with the copper centre through a hydrogen bond at the NH2 group of aspartate ion. It is important to notice that this N-terminal BSA site is also a model for the synthesized amidate tripeptide, although in the model the NH2 group was exchanged by a methyl group. In addition, the N-terminal was studied with one negative charge and total spin ½ ħ.
Discussion
Earlier studies in the literature described a characteristic negative/positive pattern in the visible range of the CD spectra of albumin solutions when incremental additions of ca. 1 mol equivalent of copper(II) ions (as the aqua-complex) were made. For BSA, bands at 556 and 475 nm were attributed to d-d transitions of the copper(II) ion, and that at 311 nm to a ligand to metal charge transfer (LMCT) transition between a deprotonated peptide nitrogen and copper(II).8,9 Further addition of copper, up to 2 mol equivalent, led to the appearance of a novel positive band at 635 nm, but addition of the third or further mol equivalent of copper(II) ions did not affect the CD spectra. These studies indicated that probably only two sites in BSA involve copper(II) binding specific enough to produce chirality transfer from the protein to the metal ion. Further, they pointed out that the differences in affinity between the binding sites for copper are so high that the second site is populated only after saturation of the first one. The primary strong binding site was identified as the N-terminal sequence Asp-Thr-His, involving two nitrogen donors (2N) from Asp and Thr residues, for which bands at 564 and 490 nm were assigned. For the second site, also characterized by CD bands and EPR parameters, a tetragonal binding geometry involving [2N, 4O] coordination was suggested.9
Our results are indicative of different sites for the interaction of these diimine copper(II) complexes 1 to 6 with BSA, and consequent copper loading into the protein. In this aspect, they are consistent with those earlier data. Additionally, they also show a crucial influence of the ligand in defining the preferential metal binding site, since stable diimine ligands led to different interactions with at least two competitive sites, depending on the ligand and on the [CuL]/[BSA] ratio. Indeed, for one of the complexes, complex 1, only the first site seems to be available, independently of the [CuL]/[BSA] ratio used.
The results from stability constant determination indicated that the complexes are quite stable under biological conditions with relative constants very similar to that of the species [Cu(BSA)] itself,7,8 and were corroborated by the DFT calculations of binding energies.
Among the most reactive compounds, [Cu(apyepy)(OH)]+ (1), [Cu(apzhist)(OH)]+ (3) and [Cu(apzpn)]2+ (4) exhibited large amounts of carbonyl groups formed, and they predominantly loaded the primary copper binding site in BSA. However, a dissimilar reactivity of copper ions inserted in both sites of the protein was verified, depending on the imine ligand. In the case of complex [Cu(apzpn)]2+ (4), the indubitable simultaneous insertion of the metal still coordinated to the ligand in the protein18 seems to promote additional formation of ROS and, consequently, causes more extended damage to the protein, probably leading to degradation (besides carbonyl groups formation).
In a previous work, the capability of these copper(II) complexes in generating hydroxyl radicals was shown to be dependent on their redox potential, with E1/2 values in the range 0.15 to -0.20 V vs. SHE.19 Further, some of them could be considered as good SOD mimics in comparison to other examples in the literature.18,36 They were capable of dismutating superoxide radicals, specially compounds 1, 3 and 4, exhibiting IC50 in the range 10-7-10-8 mol L-1. However, the BSA carbonyl formation seems to be hardly influenced by this antioxidant property. The results pointed to a much lower or no influence of superoxide anions dismutation in the process of protein oxidation. Analogous results were obtained when the substrate was the synthesized Asp-Thr-His-NH2 tripeptide.
These data can be indicative of specific requirements for significant oxidative damage to the protein, that is, the availability of the reduced form of the complex (species being able to be reduced by hydrogen peroxide or by certain groups in the protein), and the possibility of being re-oxidized by molecular oxygen or the peroxide itself. The specificity of the copper binding site in the protein seems not to be very important for oxidative damage, since the most reactive complexes, 1, 3 and 4, showed different behavior and preference regarding copper loading to different sites in the protein. Also, it was noticed that the complexes with pyrazine groups, such as [Cu(apzhist)(OH)]+ (3) or [Cu(apzpn)]2+ (4), which are capable of extra interactions with the protein through hydrogen bonding, are among the most reactive. Indeed, our data also pointed to the possibility of linking of the copper ion to the metal binding site of the protein when still coordinated to the imine ligand. This is particularly significant, and was undoubtedly proved, for complex 4, but can also be considered for the other complexes, especially at the second binding site. For complex 6, however, the reactivity was lower than the expected considering its structural features (presence of imidazole moieties). Further studies are needed in order to better identify the second metal binding site in the protein targeted by these copper complexes, and are in development in our laboratory.The calculated binding energies for all the Schiff bases are similar. The smallest and largest values were determined for complexes 2 and 6 respectively, which differ by 42 kcal mol-1. The higher stability exhibited by complex 6 can be attributed to the small bond distances to the copper(II) ion in the imidazole moieties (Figure 7). The [Cu(BSA)]+ species is more stable than all the other complexes since the negatively charged ligand (N-terminal) provides an additional stabilization through electrostatic interactions with copper(II). The deformation energies are related to the ligand features, and as expected tetradentate species are more distorted than the tridentate ones. This is due to the fact that a tetradentate ligand presents more steric effects and a more remarkable tetrahedral distortion. The large value of ΔEdef observed at the N-terminal site is attributed to its necessary structural changes in order to acquire an adequate conformation for coordination to copper(II). On comparing the relative stability constants (actually log KCuL) with ΔEb for all the Schiff base complexes, it is clear that the values of log KCuL are very similar (12.6 to 14.0 ± 0.5), differing by at most 10%, which is the same variation observed among the calculated binding energies.
The molecular graphs shown in Figure 7 are in agreement with the Poincaré-Hopf rule,42 indicating not only that the obtained complexes are well stabilized in terms of electron density distribution, but also pointing out that the free bases can be stabilized by intramolecular and hydrogen bonds, for example in apzhist, apzpn and dachist. The calculated data suggest that all Cu-N and Cu-O bonds have a strong ionic character, as indicated by the positive values of the Laplacian (∇2ρb > 0), whose values range from +0.227 up to +0.322 (a.u.). This indicates that the ligands employed are quite similar, and that they do not affect remarkably the charge distribution or the charge polarization around the metal centre. Complex 6 presents the most positive values of ∇2ρb (around 0.347 a.u.) for Cu-N bonds, indicating that, in this case, Cu-N bonds are more polarized. Since the Cu-O and Cu-N bonds have a strong ionic character, their densities values at BCPs are consequently very small, in comparison to ordinary covalent bonds. The small density and positive Laplacian values at BCPs result from the fact that the electron density contracts away from the contact region of the interacting atoms. For Cu-N or Cu-O bonds, independent of the ligand, the values of Gb and |Vb| are almost comparable, but |Vb| is rather larger than Gb (see Supporting Information - Table S1).
The topological results reinforce the fact that the copper(II) ion is better stabilized at the N-terminal site of BSA, in which the Laplacian presented negative values for Cu-N bonds and a slightly covalent character (as indicated by the less positive values of Hb), in comparison with the imine complexes (Table S1). For [Cu(BSA)]+, the C-N bonds become much more polarized than in complexes 1-6, exhibiting a slightly gain of covalence. The Cu-N polarization is related with the increase of the nitrogen nucleophilicity, which is directly affected by the presence of intramolecular hydrogen bonds, as shown in Figure 7.
Conclusions
By using spectroscopic techniques, CD, EPR and UV-Vis, different binding sites for diimine copper complexes were observed in BSA. Most of the complexes interacted first at the primary binding site of the protein, assigned as the square planar N-terminal site, up to the ratio [CuL]/[BSA] = 1:1, and only at higher ratios loaded a second metal binding site. However, one of them, complex 1, exhibited copper binding exclusively at the primary site. The relative thermodynamic formation constants of these copper(II) complexes were determined in competitive equilibria with BSA, and seem to be very similar to that of the copper-protein species itself, with KCuL values in the range of 4×1012 to 1×1014. In all cases, some evidence for simultaneous coordination of the copper ion to the imine ligand and binding groups in the protein was detected, especially for complex 4. The redox potential for these complexes was verified in a quite sharp range (0.15 to -0.20 V vs. SHE) barely related to the ligand features,18 and so cannot entirely explain the observed differences in reactivity.
The obtained binding energies for the copper ion coordinated to each of these ligands were very similar, and not very far from that determined for the N-terminal metal binding site of the protein, attesting the high stability of copper when inserted into this kind of environment.
The more reactive complexes towards both the BSA and the tripeptide oxidation were complexes [Cu(apyepy)(OH)]+ (1) > [Cu(apzhist)(OH)]+ (3) ≈ [Cu(apzpn)]2+ (4), which also showed high capability of generating hydroxyl radicals, suggesting their participation in monoelectronic pathways, in a predominant radicalar mechanism. On the other hand, the verified SOD activity of these complexes seems not to influence the protein oxidation. Therefore, superoxide anions do not play a relevant role in this process. In the case of complex 4, an extra or alternative degradative oxidation of the protein probably occurs, since the amount of carbonyl groups formed is much lower than that expected from its capacity of ROS formation. The data obtained with the synthetic tripeptide Asp-Thr-His-NH2 complemented the results obtained with the protein, and helped on getting a better characterization of its metal binding sites.
Supplementary Information
Supplementary data is available free of charge at http://jbcs.sbq.org.br, as a PDF file.
Acknowledgments
Financial support by the Brazilian agencies FAPESP (grant 05/60596-8), CNPq and CAPES is gratefully acknowledged. A. M. D. C. F. was also supported by the Programa Institutos Nacionais de Ciência e Tecnologia - CNPq/FAPESP/MCT (INCT Processos Redox em Biomedicina - Redoxoma). M. P. A. thanks CAPES for a doctoral fellowship; M. A. A. A. and A. M. were recipients of post-doctoral fellowships from FAPESP. We also thank Dr. Adelaide Faljoni-Alario for helping on CD measurements. G. F. C. thanks CENAPAD-SP for the excellent service and computational time provided.
References
1. Carter, D. C.; Ho, J. X.; Adv. Protein Chem. 1994, 45, 153.
2. Sadler, P. J.; Tucker, A.; Viles, J. H.; Eur. J. Biochem. 1994, 220, 193.
3. Masuoka, J.; Hegenauer, J; Van Dyke, B. R.; Saltman, P.; J. Biol. Chem. 1993, 268, 21533.
4. Ivanov, A. I.; Parkinson, J. A.; Cossins, E.; Woodrow, J.; Sadler, P. J.;. J. Biol. Inorg. Chem. 2000, 5, 102.
5. Zgirski, A.; Frieden, E.; J. Inorg . Biochem. 1990, 39, 137.
6. Zhang, Y.; Wilcox, D. E.; J. Biol. Inorg. Chem. 2002, 7, 327.
7. Masuoka, J.; Saltman, P.; J. Biol. Chem. 1994, 269, 25557.
8. Rózga, M.; Sokolowska, M.; Protas, A. M.; Bal, W.; J. Biol. Inorg. Chem. 2007, 12, 913.
9. Bal, W.; Christodoulou, J.; Sadler, P. J.; Tucker, A.; J. Inorg. Biochem. 1998, 70, 33.
10. Shrivastava, H. Y.; Kanthimathi, M.; Nair, B. U.; Biochim. Biophys. Acta 2002, 1573, 149.
11. Boghaei, D. M.; Farvid, S. S.; Gharagozlou, M.; Spectrochim. Acta Part A 2007, 66, 650.
12. Kito, M.; Takenaka, Y.; Urade, R.; FEBS Lett. 1995, 362, 39.
13. Nishida, Y.; Takeuchi, M.; Oishi, N.; Kida, S.; Inorg. Chim. Acta 1985, 96, 81.
14. Valentine, J. S.; Hart, P. J.; Proc. Natl. Acad. Sci USA 2003, 100, 3617.
15. Bush, A. I.; Trends Neurosci. 2003, 26, 207.
16. Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G.; Chem. Rev. 2006, 106, 1995.
17. Silveira, V. C.; Caramori, G. F.; Abbott, M. P.; Gonçalves, M. B.; Petrilli, H. M.; Ferreira, A. M. D. C.; J. Inorg. Biochem. 2009, 103, 1331.
18. Santos, M. L. P.; Faljoni-Alario, A.; Mangrich, A. S.; Ferreira, A. M. D. C.; J. Inorg. Biochem. 1998, 71, 71.
19. Azzellini, M. A. A.; Bagatin, I. A.; Ferreira, A. M. D. C.; Redox Report 2006, 11, 25.
20. Opazo, C.; Barria, M. I.; Ruiz, F. H.; Inestrosa, N. C.; Biometals 2003, 16, 91.
21. For example: Cerchiaro, G.; Ferreira, A. M. D. C.; J. Braz. Chem. Soc. 2006, 17, 1473, and references cited herein.
22. Ellman, G.; Lysko, H.; Anal. Biochem. 1979, 93, 98.
23. Isles, T. E.; Jocelyn, P. C.; Biochem. J. 1963, 88, 84.
24. Buettner, G. R.; Oberley, L. W.; Biochem. Biophys. Res. Commun. 1978, 8, 69.
25. Nogueira, R. F. P.; Oliveira, M. C.; Paterlini, W. C.; Talanta 2005, 66, 86.
26. Vigato, P. A.; Tamburini, S.; Coord. Chem. Rev. 2004, 248, 1717.
27. Santos, M. L. P.; Bagatin, I. A.; Pereira, E. M.; Ferreira, A. M. D. C. ; J. Chem. Soc., Dalton Trans. 2001, 838.
28. Stewart, J.; Young, J. D.; Solid Phase Peptide Synthesis, 2nd ed., Pierce Chemical Company: Rockford, IL, 1984, pp. 1-49.
29. Varanda, L. M.; Miranda, M. T. M.; J. Peptide Res. 1997, 50, 102.
30. Souza, M. P.; Tavares, M. F. M.; Miranda, M. T. M.; Tetrahedron 2004, 60, 4671.
31. Kaiser, E.; Colescott, R. L.; Bossinger, C. D.; Cook, P. I.; Anal. Biochem. 1970, 34, 595.
32. Tordo, P.; Electron Paramagnetic Resonance (A Specialist Periodical Report), Royal Soc. Chem.: Cambridge, UK, 1998, 16, pp. 116-144.
33. Reznick, A. Z.; Packer, L.; Methods Enzymol. 1994, 233, 357.
34. Hawkins, C. L.; Davies, M. J.; Biochim. Biophys. Acta 1997, 1360, 84.
35. Beauchamp, C.; Fridovich, I.; Anal. Biochem. 1971, 44, 276.
36. Müller, J.; Felix, K.; Maichle, C.; Lengfelder, E.; Strähle, J.; Weser, U.; Inorg. Chim. Acta 1995, 233, 11.
37. Hassan, H. M.; Free Radical Biol. Med. 1988, 5, 377.
38. Blöchl, P. E.; Phys. Rev. B 1994, 50, 17953.
39. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.: Gaussian 03, Revision C. 02, Gaussian, Inc., Pittsburgh PA, 2003.
40. Blöchl, P. E.; J. Chem. Phys. 1995, 103, 7422.
41. Perdew, J. P.; Burke K.; Ernzerhof, M.; Phys. Rev. Lett. 1996, 77, 3865.
42. Bader, R. F.; Atoms in Molecules. A Quantum Theory, Claredon: Oxford, 1990; Popelier, P. L. A.; Atoms in Molecules - An Introduction, 1st ed., Prentice Hall: England, U.K., 2000; Several Contributors in The Quantum Theory of Atoms in Molecules; Matta, C. F.; Boyd, R. J., ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2007.
43. Becke, A. D.; Phys. Rev. A. 1988, 38, 3098; Perdew, J. P.; Phys. Rev. B. 1986, 33, 8822.
44. Schaefer, A.; Horn, H.; Ahlrichs, R.; J. Chem. Phys. 1992, 97, 2571; Schaefer, A.; Huber, C.; Ahlrichs, R.; J. Chem. Phys. 1994, 100, 5829.
45. Bader, R. F. W. (1995) AIMPAC - Source code obtained from the AIMPAC site at http://www.chemistry.mcmaster.ca/aimpac/aimpac.html, McMaster University, Hamilton, USA.
46. Bianchi, R.; Gervasio, G.; Marabello, D.; Inorg. Chem. 2000, 39, 2360.
47. Wardell, M.; Wang, Z.; Ho, J. X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D. C.; Biochem. Biophys. Res. Comm. 2002, 291, 813.
48. Panwen, S.; Yongqia Z.; Sheyi, W.; Yunxia, C.; Inorg. Chim. Acta 1990, 169, 161.
Received: October 2, 2009
Web Release Date: April 22, 2010
FAPESP helped in meeting the publication costs of this article.
Supplementary Information
- 1. Carter, D. C.; Ho, J. X.; Adv. Protein Chem 1994, 45, 153.
- 2. Sadler, P. J.; Tucker, A.; Viles, J. H.; Eur. J. Biochem 1994, 220, 193.
- 3. Masuoka, J.; Hegenauer, J; Van Dyke, B. R.; Saltman, P.; J. Biol. Chem 1993, 268, 21533.
- 4. Ivanov, A. I.; Parkinson, J. A.; Cossins, E.; Woodrow, J.; Sadler, P. J.;. J. Biol. Inorg. Chem 2000, 5, 102.
- 5. Zgirski, A.; Frieden, E.; J. Inorg . Biochem 1990, 39, 137.
- 6. Zhang, Y.; Wilcox, D. E.; J. Biol. Inorg. Chem 2002, 7, 327.
- 7. Masuoka, J.; Saltman, P.; J. Biol. Chem 1994, 269, 25557.
- 8. Rózga, M.; Sokolowska, M.; Protas, A. M.; Bal, W.; J. Biol. Inorg. Chem 2007, 12, 913.
- 9. Bal, W.; Christodoulou, J.; Sadler, P. J.; Tucker, A.; J. Inorg. Biochem. 1998, 70, 33.
- 10. Shrivastava, H. Y.; Kanthimathi, M.; Nair, B. U.; Biochim. Biophys. Acta 2002, 1573, 149.
- 11. Boghaei, D. M.; Farvid, S. S.; Gharagozlou, M.; Spectrochim. Acta Part A 2007, 66, 650.
- 12. Kito, M.; Takenaka, Y.; Urade, R.; FEBS Lett. 1995, 362, 39.
- 13. Nishida, Y.; Takeuchi, M.; Oishi, N.; Kida, S.; Inorg. Chim. Acta 1985, 96, 81.
- 14. Valentine, J. S.; Hart, P. J.; Proc. Natl. Acad. Sci USA 2003, 100, 3617.
- 15. Bush, A. I.; Trends Neurosci. 2003, 26, 207.
- 16. Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G.; Chem. Rev. 2006, 106, 1995.
- 17. Silveira, V. C.; Caramori, G. F.; Abbott, M. P.; Gonçalves, M. B.; Petrilli, H. M.; Ferreira, A. M. D. C.; J. Inorg. Biochem 2009, 103, 1331.
- 18. Santos, M. L. P.; Faljoni-Alario, A.; Mangrich, A. S.; Ferreira, A. M. D. C.; J. Inorg. Biochem 1998, 71, 71.
- 19. Azzellini, M. A. A.; Bagatin, I. A.; Ferreira, A. M. D. C.; Redox Report 2006, 11, 25.
- 20. Opazo, C.; Barria, M. I.; Ruiz, F. H.; Inestrosa, N. C.; Biometals 2003, 16, 91.
- 21. For example: Cerchiaro, G.; Ferreira, A. M. D. C.; J. Braz. Chem. Soc 2006, 17, 1473, and references cited herein.
- 22. Ellman, G.; Lysko, H.; Anal. Biochem 1979, 93, 98.
- 23. Isles, T. E.; Jocelyn, P. C.; Biochem. J. 1963, 88, 84.
- 24. Buettner, G. R.; Oberley, L. W.; Biochem. Biophys. Res. Commun. 1978, 8, 69.
- 25. Nogueira, R. F. P.; Oliveira, M. C.; Paterlini, W. C.; Talanta 2005, 66, 86.
- 26. Vigato, P. A.; Tamburini, S.; Coord. Chem. Rev 2004, 248, 1717.
- 27. Santos, M. L. P.; Bagatin, I. A.; Pereira, E. M.; Ferreira, A. M. D. C. ; J. Chem. Soc., Dalton Trans. 2001, 838.
- 28. Stewart, J.; Young, J. D.; Solid Phase Peptide Synthesis, 2nd ed., Pierce Chemical Company: Rockford, IL, 1984, pp. 1-49.
- 29. Varanda, L. M.; Miranda, M. T. M.; J. Peptide Res 1997, 50, 102.
- 30. Souza, M. P.; Tavares, M. F. M.; Miranda, M. T. M.; Tetrahedron 2004, 60, 4671.
- 31. Kaiser, E.; Colescott, R. L.; Bossinger, C. D.; Cook, P. I.; Anal. Biochem. 1970, 34, 595.
- 32. Tordo, P.; Electron Paramagnetic Resonance (A Specialist Periodical Report), Royal Soc. Chem.: Cambridge, UK, 1998, 16, pp. 116-144.
- 33. Reznick, A. Z.; Packer, L.; Methods Enzymol. 1994, 233, 357.
- 34. Hawkins, C. L.; Davies, M. J.; Biochim. Biophys. Acta 1997, 1360, 84.
- 35. Beauchamp, C.; Fridovich, I.; Anal. Biochem. 1971, 44, 276.
- 36. Müller, J.; Felix, K.; Maichle, C.; Lengfelder, E.; Strähle, J.; Weser, U.; Inorg. Chim. Acta 1995, 233, 11.
- 37. Hassan, H. M.; Free Radical Biol. Med. 1988, 5, 377.
- 38. Blöchl, P. E.; Phys. Rev. B 1994, 50, 17953.
- 39. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.: Gaussian 03, Revision C. 02, Gaussian, Inc., Pittsburgh PA, 2003.
- 40. Blöchl, P. E.; J. Chem. Phys 1995, 103, 7422.
- 41. Perdew, J. P.; Burke K.; Ernzerhof, M.; Phys. Rev. Lett 1996, 77, 3865.
- 42. Bader, R. F.; Atoms in Molecules. A Quantum Theory, Claredon: Oxford, 1990;
- Popelier, P. L. A.; Atoms in Molecules - An Introduction, 1st ed., Prentice Hall: England, U.K., 2000;
- Several Contributors in The Quantum Theory of Atoms in Molecules; Matta, C. F.; Boyd, R. J., ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2007.
- 43. Becke, A. D.; Phys. Rev. A. 1988, 38, 3098;
- Perdew, J. P.; Phys. Rev. B. 1986, 33, 8822.
- 44. Schaefer, A.; Horn, H.; Ahlrichs, R.; J. Chem. Phys. 1992, 97, 2571;
- Schaefer, A.; Huber, C.; Ahlrichs, R.; J. Chem. Phys. 1994, 100, 5829.
- 45. Bader, R. F. W. (1995) AIMPAC - Source code obtained from the AIMPAC site at http://www.chemistry.mcmaster.ca/aimpac/aimpac.html, McMaster University, Hamilton, USA.
- 46. Bianchi, R.; Gervasio, G.; Marabello, D.; Inorg. Chem. 2000, 39, 2360.
- 47. Wardell, M.; Wang, Z.; Ho, J. X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D. C.; Biochem. Biophys. Res. Comm. 2002, 291, 813.
- 48. Panwen, S.; Yongqia Z.; Sheyi, W.; Yunxia, C.; Inorg. Chim. Acta 1990, 169, 161.
Publication Dates
-
Publication in this collection
30 July 2010 -
Date of issue
2010
History
-
Received
02 Oct 2009 -
Accepted
22 Apr 2010