Abstracts
A new catalytic route with potential practical interest to sustainable production of bio-additives from glycerol is described. Ethyl acetate was transesterified with glycerol, in the ratio glycerol:EtOAc 1:10, at 25 or 90 ºC using 0.1 equiv. of H2SO4 or TsOH, as homogeneous catalysts. H2SO4 led to the total glycerol consumption in 2 h. In the equilibrium, attained in 9 h, 100% yield of a diacetin:triacetin (55:45) mixture was formed. Using AmberlystTM 15 dry and AmberlystTM 16 wet in 1:30 glycerol:EtOAc ratio and reflux at 90 ºC the total glycerol consumption was achieved in 2 and 10h, respectively. The lower reactivity of Amberlyst-16 wet was explained in terms of deactivation of acid sites and decrease in glycerol diffusion to the inner resin pores, both factors caused by adsorbed water. The kinetics of glycerol transformation and product distribution in the equilibrium in relation to the H2SO4, Amberlyst-15 (dry) and Amberlyst-16 (wet) catalyzed reactions were measured.
triacetin; diacetin; Amberlyst-16 wet; acidic ion-exchange resins; glycerol acetylation
Uma nova rota catalítica, de potencial interesse prático para a produção sustentável de acetinas a partir do glicerol, é descrita. Acetato de etila foi transesterificado com glicerol, numa razão glicerol:EtOAc 1:10, a 25 ou 90 ºC, usando 0,1 equivalente de H2SO4 ou TsOH como catalisadores homogêneos. H2SO4 levou ao consumo total de glicerol, a 90 ºC, em 2 h. No equilíbrio, atingido em 9 h, 100% de uma mistura diacetina:triacetina (55:45) foi formada. Usando-se AmberlystTM 15 seca e AmberlystTM 16 úmida numa razão glicerol:EtOAc 1:30 sob refluxo a 90 ºC, obteve-se o consumo total do glicerol em 2 e 10 h, respectivamente. A menor reatividade da resina Amberlyst-16 úmida foi explicada pela desativação dos sítios ácidos e pela diminuição da difusão do glicerol para o interior dos poros, causada pela água adsorvida. A cinética da transformação do glicerol e a distribuição de produtos no equilíbrio para a reação catalisada por H2SO4, AmberlystTM 15 seca e AmberlystTM 16 úmida foram medidas e racionalizadas.
ARTICLE
Synthesis of bio-additives: transesterification of ethyl acetate with glycerol using homogeneous or heterogeneous acid catalysts
Bruno A. Meireles; Vera Lúcia P. Pereira* * e-mail: patrocinio@nppn.ufrj.br
Núcleo de Pesquisas de Produtos Naturais, Centro de Ciências da Saúde, Universidade Federal do Rio de Janeiro, Bloco H, Cidade Universitária, 21941-902 Rio de Janeiro-RJ, Brazil
ABSTRACT
A new catalytic route with potential practical interest to sustainable production of bio-additives from glycerol is described. Ethyl acetate was transesterified with glycerol, in the ratio glycerol:EtOAc 1:10, at 25 or 90 ºC using 0.1 equiv. of H2SO4 or TsOH, as homogeneous catalysts. H2SO4 led to the total glycerol consumption in 2 h. In the equilibrium, attained in 9 h, 100% yield of a diacetin:triacetin (55:45) mixture was formed. Using AmberlystTM 15 dry and AmberlystTM 16 wet in 1:30 glycerol:EtOAc ratio and reflux at 90 ºC the total glycerol consumption was achieved in 2 and 10h, respectively. The lower reactivity of Amberlyst-16 wet was explained in terms of deactivation of acid sites and decrease in glycerol diffusion to the inner resin pores, both factors caused by adsorbed water. The kinetics of glycerol transformation and product distribution in the equilibrium in relation to the H2SO4, Amberlyst-15 (dry) and Amberlyst-16 (wet) catalyzed reactions were measured.
Keywords: triacetin, diacetin, Amberlyst-16 wet, acidic ion-exchange resins, glycerol acetylation
RESUMO
Uma nova rota catalítica, de potencial interesse prático para a produção sustentável de acetinas a partir do glicerol, é descrita. Acetato de etila foi transesterificado com glicerol, numa razão glicerol:EtOAc 1:10, a 25 ou 90 ºC, usando 0,1 equivalente de H2SO4 ou TsOH como catalisadores homogêneos. H2SO4 levou ao consumo total de glicerol, a 90 ºC, em 2 h. No equilíbrio, atingido em 9 h, 100% de uma mistura diacetina:triacetina (55:45) foi formada. Usando-se AmberlystTM 15 seca e AmberlystTM 16 úmida numa razão glicerol:EtOAc 1:30 sob refluxo a 90 ºC, obteve-se o consumo total do glicerol em 2 e 10 h, respectivamente. A menor reatividade da resina Amberlyst-16 úmida foi explicada pela desativação dos sítios ácidos e pela diminuição da difusão do glicerol para o interior dos poros, causada pela água adsorvida. A cinética da transformação do glicerol e a distribuição de produtos no equilíbrio para a reação catalisada por H2SO4, AmberlystTM 15 seca e AmberlystTM 16 úmida foram medidas e racionalizadas.
Introduction
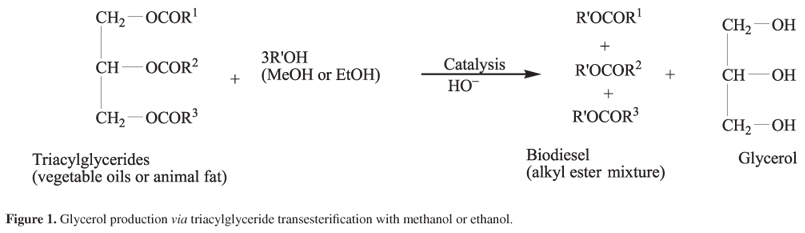
Biodiesel is usually produced via base catalyzed transesterification reaction between methanol or ethanol and triacylglycerides. Glycerol is the main co-product from this transformation. Its production is equivalent to approximately 10 wt% of the total biodiesel production (Figure 1).1-3
Glycerol offers a variety of applications in different areas.4 For example, it has been used as a humectant, emollient, demulcent, solvent, and thickener in food and pharmaceutical manufacturing. Other uses include production of explosives, dyes, polymers, lubricants, printing ink and papers in the chemical industry.5-8 More recently it has been used as a building block and solvent in organic synthesis.9-11 Biologically, it is currently used as a cryoprotectant of different biological materials, in purification of proteins and enzymes, in bacterial cultures, and in the production of bio-plastics, among other uses.12 Despite its enormous industrial applicability, the predictably increasing volumes of glycerol generated from biodiesel production will create a glut in the market in the short term. It is estimated that the worldwide production of glycerol in 2012 will be around 1.2 million tonnes.6 To make biodiesel economically viable, new and efficient routes for transforming glycerol into higher value products must be developed.5-8 Although most biodiesel physico-chemical properties have close resemblance to petrol diesel, freezing problems and increase in viscosity at low temperatures, besides low resistance to oxidation, render it inferior in comparison to petrol diesel. The addition of bio-additives derived from glycerol could lead to an improvement in biodiesel efficiency and glycerol valorization, thus decreasing the total cost biodiesel production making it an even more attractive fuel.13,14 Several additives such as ethers,15,16 carbonates,17 acetals18,19 and esters of glycerol13 have already been synthesized. In this context, the production of acetins is a chemically suitable application for the excess glycerol from biodiesel production.20-30 Acetins are mono-, di-, and triacetate esters of glycerol. Monoacetins and diacetins have wide applications as raw materials for biodegradable polyesters, pharmaceuticals, food and cosmetic industries.20,21 Triacetin occupies 10% of the worldwide glycerol market as fuel additives, antiknock additive for gasoline, additives in cosmetics, in foods, and as solvents, among other uses.22,23 Conventionally, acetins are synthesized via glycerol esterification with excess acetic acid and/or acetic anhydride under mineral acid catalysis.24 Currently, heterogeneous catalysts such as acidic ion-exchange organic resins,20,21 zeolites,25 niobic acid,26 activated carbon-supported heteropolyacids,27 zirconia-based catalysts,28 metal/SBA-15 catalyst29 among others are being utilized in the synthesis of acetins. Despite being widely used the acid catalyzed esterification process suffers from drawbacks such as high costs, corrosive/toxic reagents' use leading to environment problems and economical inefficiency.30
We have investigated another possible route for acetin production consisting of transesterification of ethyl acetate, using homogeneous or heterogeneous acid catalysis.31,32 To the best of our knowledge, no systematic study for glycerol acetylation via transesterification of ethyl acetate or other alkyl esters had been performed prior to ours. In the course of the writing this work, Morales et al.33 described a second study where the transesterification of methyl acetate with glycerol catalyzed by different heterogeneous catalysts was investigated.
We wish to report here our detailed results about the production of acetins via the transesterification reaction of ethyl acetate with glycerol using sulfuric acid (H2SO4), p-toluenesulfonic acid (TsOH) or piridinium p-toluenesulfonate (PPTS) as homogeneous catalysts and Amberlyst-15 (dry) resin or Amberlyst-16 (wet) resin as heterogeneous catalysts (Figure 2).
A study on the influence of parameters such as temperature effect, molar ratio of glycerol:ethyl acetate and catalyst loading was accomplished. In addition, the equilibrium selectivity was determined for the most efficient catalysts.
Experimental
Materials
Glycerol (99.5%) and ethyl acetate (99.5%) both p.a.-ACS were used as well as sulfuric acid, 96% (H2SO4), p-toluenesulfonic acid (PTSA) and pyridinium p-toluenesulfonate (PPTS), all purchased from Vetec Química Fina Ltda and used as provided. AmberlystTM 15 dry and AmberlystTM 16 wet were purchased from Sigma-Aldrich® and readily utilized. The catalyst properties were considered as informed in the specifications provided by the manufacturer (Rohm and Haas Company).
Methods
Gas chromatography analyses were performed every 60 min, using a GC-FID (Shimadzu GC-FID 2010; DB-1MS (100% polydimethylsiloxane) fused silica capillary column (30 m × 0.25 mm, film thickness 0.25 µm); carrier gas H2 (1.0 mL min-1); temperature: injector 290 ºC, column oven 60-290 ºC at 10 ºC min-1, FID 290 ºC). Acetins were the only detected products exhibiting the retention times of 6.16 min (monoacetin); 8.13 min (diacetin); 9.51 min (triacetin). These times were obtained by GC from isolated and characterized samples.32 Acetin conversion was measured relative to glycerol absolute conversion (retention time 4.92 min); 1H NMR and 13C NMR spectra were recorded on Varian MR-400 (400 MHz) with TMS as internal reference. The coupling constant (J) is in hertz (Hz). IR spectra were recorded on a Shimadzu IR-Prestige-21 (FTIR) spectrophotometer as a film on NaCl plate. Analyses by GC-MS were performed on a Shimadzu GC/MS-QP 500.
Acetylation reactions in homogeneous media
All reactions were carried out in batch mode. Glycerol (5.0 g, 54.3 mmol, 1.0 equiv.), ethyl acetate (47.8 g, 60.6 mL, 543 mmol, 10 equiv.) and an acid catalyst (5.43 mmol, 0.1 equiv.) were added to a round bottom flask. Next, a Liebig condenser and a pressure equalizer were adapted to the system and the reaction medium was kept under strong magnetic stirring at room temperature or 90 ºC for 24 h. Initially, two phases were formed but at 45 min (90 ºC) and 180 min (room temperature) only one phase could be observed. Next, the reaction medium was diluted with ethyl acetate (500 mL) washed with saturated NaHCO3 (3 × 20.0 mL) and brine (1 × 15 mL). The organic phase was dried over Na2SO4 and the solvent removed in vacuum. The residue was constituted of a low viscosity yellow liquid consisting of an acetin mixture (100% conversion, 10.1 g of acetins). Acetins separation could be done by column chromatography on silica gel (hexane/EtOAc 4:1) or more easily by counter current chromatograph resulting in colorless liquids.32
Spectroscopy data for monoacetin (mixture of 1:2-regioisomers): 1H NMR (400 MHz, CDCl3) δ 2.10 (m, 6H), 3.67 (brs, 2H), 3.11(brs, 1H), 3.00 (brs, 1H), 4.14-4.02 (m, 8H), 4.25 (m, 1H), 5.01 (m, 1H); 13C NMR (101 MHz, CDCl3) δ ppm 20.6, 20.9, 61.1, 62.3, 65.1, 67.8, 72.2, 170.6, 171.1; GC-MS (70 eV) m/z 43, 103, 73; IR (film) νmax/cm-1: 1743, 1367, 1240, 1050.
Spectroscopy data for diacetin (2.8:1 mixture of 1,3:1,2-regioisomers): 1H NMR (400 MHz, CDCl3) δ 2.08 (m,12H), 4.36-4.01 (m, 5H), 4.67 (brs,1H), 5.07 (m,1H); 13C NMR (100 MHz, CDCl3) δ 19.9, 20.1, 59.9, 61.7, 62.2, 66.6, 71.4, 170.0, 170.4; GC-MS (70 eV) m/z 43, 103, 73; IR (film) νmax/cm-1: 1740, 1373, 1233.
Spectroscopy data for triacetin: 1H NMR (400 MHz, CDCl3) δ 2.03 (d, 9 H, J 8Hz), 4.1 (dd, 2H, J 4.0, 8.0 Hz), 4.2 (dd, 2H, J 8.0, 8.0 Hz), 5.2 (m, 1H), 4.2 (dd, 2H, J 8.0, 8.0 Hz), 5.2 (m,1H); 13C NMR (101 MHz, CDCl3) δ 20.5, 20.6, 62.1, 68.9, 169.9, 170.3; CG/MS (70 eV) m/z 43, 145, 103, 73; IR (film) νmax/cm-1: 1747, 1372, 1225, 1501.
Acetylation reactions in heterogeneous media
All reactions were carried out in batch mode. Glycerol (1.0 g, 10.9 mmol, 1.0 equiv.), ethyl acetate (327 mmol, 28.8 g, 36.5 mL, 30 equiv.) and an acid catalyst (1.09 mmol, 0.1 equiv., 0.23 g of AmberlystTM 15 dry) were added to a round bottom flask. Next, a Liebig condenser and a pressure equalizer were adapted to the reaction system and the resulting triphasic mixture was magnetically stirred at room temperature for 24 h or at 90 ºC for 18 h. After 1.5 h it was possible to observe a biphasic mixture. Complete glycerol consumption was reached in 2 and 10 h when Amberlyst-15 (dry) and Amberlyst-16 (wet) acid resins were used, respectively. Simple filtration, washing with ethyl acetate (twice the reaction volume) and removal of volatile portions under reduced pressure led to a colorless liquid of low viscosity comprising a mixture of acetins (1.9 g).
Results and Discussion
After several trials it was discovered that the 1:10 glycerol:EtOAc ratio, under strong magnetic stirring, using 0.1 equiv. of homogeneous catalyst, and at room temperature allows an adequate fluidity of the reaction medium, with one phase being observed after 3 h of reaction. Thus, this ratio was utilized in the experiments that employed homogeneous catalysts. On the other hand, the use of a stoichiometric 1:3 glycerol:EtOAc ratio produced an extremely viscous medium and a low glycerol conversion (ca. 20%) was obtained. Contrarily for the heterogeneous catalysts (0.1 equiv.) the use of a higher glycerol:EtOAc ratio (1:30) was necessary. In both cases, it was observed that as the reaction progresses the media becomes more fluid due to the formation of the less viscous acetins and mainly ethanol production.
From the trial results, acetin formation was investigated by reacting 1 equiv. glycerol with 10 equiv. of ethyl acetate, at room temperature, for 24 h using 0.1 equiv. of H2SO4, TsOH (p-toluenesulfonic acid), PPTs (piridinium p-toluenesulfonate), Amberlyst-15 (dry) and Amberlyst-16 (wet) acid resin as catalysts, as shown in Figure 3.
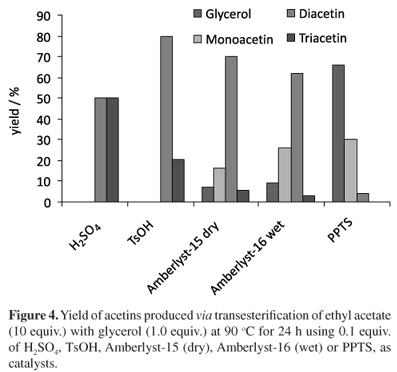
Under these conditions, it was observed that the use of H2SO4 and TsOH led to total glycerol conversion into an acetin mixture. PPTS was not reactive. Contrarily, the use of the heterogeneous catalysts Amberlyst-16 (wet) and Amberlyst-15 (dry) was less effective since 23 and 38% of glycerol, respectively, remained unreacted without observable formation of triacetin. The lower activity in solid catalysts is often related to other factors, among them external/internal diffusion that hindered the access of reagents to the active acid sites of the catalysts.34 Indeed, in the 1:10 glycerol/EtOAc ratio and at room temperature it is difficult for the reagents to access the active sites of the outer and inner surface resins because of the viscosity of the reaction medium. This mass transfer resistance is the main reason for the lower performance of the Amberlyst-15 (dry) and -16 (wet) ion exchange resins compared to homogeneous catalysts. Aiming to increase glycerol conversion for the reactions catalyzed by solid resins and PPTS besides increasing the overall triacetin production, the reactions were carried out at 90 ºC under the same reaction conditions, as shown in Figure 4.
This temperature was employed because it could lead to a lowering of the viscosity of the reaction medium and inhibits the formation of unidentified by-products which were observed when the reaction was done at 120 ºC. As expected, an increase in the temperature improved the glycerol conversion in the reactions catalyzed by PPTS, Amberlyst-16 (wet) and Amberlyst-15 (dry)34 and a general increase in the triacetin production. A mechanism based on the literature30 was proposed for homogeneous acid-catalyzed transesterification of ethyl acetate with glycerol and may explain these results, as shown in Figure 5.
The transesterification reaction involves several reversible successive steps that convert ethyl acetate into monoacetin, diacetin and triacetin, producing 1 mol of ethanol for each mol of acetin formed. The initial step is the fast protonation of the carbonyl group from ethyl acetate leading to the formation of a more electrophilic species (I). The second step, which is rate determining, consists of a nucleophilic attack of the glycerol primary hydroxyl to the activated ethoxycarbonyl group affording a tetrahedral intermediate (II) which undergoes a quick ethanol elimination to form monoacetin and regenerate the catalyst. The di and triacetin formation follows a similar process. In 2009, Granados et al.36 suggested that an analogous transesterification mechanism can also occur on a resin surface. It is worth mentioning that both monoacetin and diacetin consist of an inseparable regioisomer mixture (III + IV) and (V + VI), respectively. These regioisomers are in a dynamic equilibrium and interconvert rapidly through an internal migration of the acetyl group to the vicinal hydroxyl group and thus individual isomers cannot be isolated.32,35 In our studies it was observed that the homogeneous acidic catalysts H2SO4 and TsOH favored a higher triacetin proportion in comparison with solid acid resins Amberlyst-15 (dry) and -16 (wet), which in turn favored a higher diacetin concentration, as shown in Figures 3 and 4. This behavior is due to the influence of the acid strength on the reaction rate-determining step. Thus, H2SO4 (pka = -2) yielded a greater triacetin concentration compared to TsOH (pka = 2). PPTS (pka = 5.5), a very weak organic acid was either unreactive or poorly reactive at 90 ºC. Considering the mechanism proposed in Figure 5, triacetin production is less favored because the nucleophilic attack of the voluminous diacetin on the activated ethoxycarbonyl is more sterically hindered, resulting in a lower triacetin concentration. The use of stronger acids leads to greater concentration of H+ ions in solution and consequently, a greater concentration of electrophilic species. In turn, this greater concentration of electrophilic species favors the determining step of the reaction, increasing the overall rate at equilibrium and in consequence the triacetin concentration. Assuming that heterogeneous transesterification proceeded through a mechanism similar to the homogeneous one,36 the acid strength and/or the number of active sites on Amberlyst TM 15 dry (4.8 mmol g-1) and AmberlystTM 16 wet (4.7 mmol g-1) were not sufficient to catalyze triacetin formation and thus greater diacetin concentration was obtained. These results will be discussed ahead.
Next, the kinetics of glycerol transformation and product distribution for the reaction catalyzed by H2SO4 was investigated as shown in Figure 6.
Total glycerol consumption occurred within 2 h and the equilibrium, which consisted of a 55:45 di:triacetin ratio was reached within 9 h. In general, acid catalyzed transesterification reactions are slower than esterification reactions and perhaps this is the reason why acetins have been produced almost exclusively via glycerol esterification reactions with acetic acid and/or acetic anhydride using homogeneous and heterogeneous acid catalysts.24,25,33 Recently, Mota et al.24 performed a very efficient glycerol acetylation via an esterification process employing a 4:1 acetic anhydride:glycerol ratio and different catalysts. Triacetin was produced with 100% selectivity and total glycerol conversion, at 60 ºC, in 20 min, when zeolite Beta or K-10 Montmorillonite were used as catalysts. Reactions with acetic acid as acylating reagent (AcOH:glicerol, 4:1) under the same catalysts were also carried out. In this case, a mixture of mono, di and triacetins was obtained, in 2 h, at 120 ºC with yields over 90%. Acetin production via esterification process demands shorter reaction times and takes advantage of the use of heterogeneous catalysts.30 On the other hand, the transesterification route employs EtOAc, a reagent of lower cost and toxicity and produces EtOH as a low toxicity waste. These advantages can compensate the problems arising by homogeneous Brönsted acid catalysts in industrial transesterification processes.37
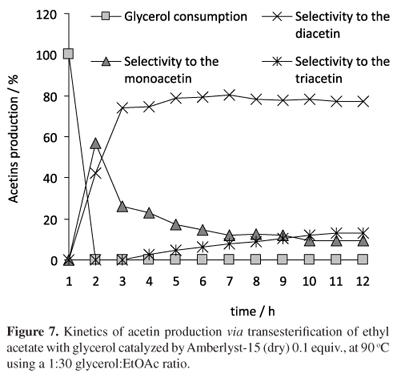
Interestingly, catalyses promoted by Amberlyst-15 (dry) and -16 (wet) acid resins (Figure 4) were quite similar to each other and led to a mixture of mono, di and triacetins with a large predominance of diacetin. Aiming to attain total glycerol conversion and the equilibrium selectivity for both resins, the reaction was carried out on Amberlyst-15 (dry) and Amberlyst-16 (wet), under the same conditions showed in Figure 4, except for the glycerol:EtOAc ratio. A dilute medium (glycerol:EtOAc, 1:30) was used to enhance stirring and total glycerol consumption and favor equilibrium. Both resins were used without any prior treatment. Figure 7 shows that Amberlyst-15 (dry) promoted total glycerol consumption in 2 h and the equilibrium was reached in approximately 20 h with a greater predominance of diacetin.
On the other hand, Amberlyst-16 (wet) led to similar products distribution in 20 h, but the total consumption of glycerol was observed in 10 h, as show in Figure 8.
Amberlyst-15 (dry) has been widely used both in industry and in academia for converting glycerol and other alcohols into high value-added products via esterification, transesterification, etherification and acetalization reactions.5,16,19 Contrarily, Amberlyst-16 (wet) has been less utilized. Usually, academic studies on its catalytic activity do not employ the resin in its wet form and water removal before using is performed.16,38,39 Amberlyst-15 (dry) and -16 (wet) are strongly acidic, macroreticular divinylbenzene copolymer matrix-based sulfonic acid resins whose main physical and structural properties are shown in Table 1.
One could expect a poor performance with Amberlyst-16 (wet) compared to Amberlyst-15 (dry) due to a higher moisture content, to an average crosslinked structure, to a smaller surface area and to a smaller macropore diameter. However, in the catalysis promoted by Amberlyst-16 (wet), total glycerol consumption was observed resulting in a product distribution very similar to that obtained with Amberlyst-15 (dry), although a longer reaction time was necessary, as shown in Figures 7 and 8. It is well documented that water reduces considerably the catalytic activity of resins and other solid acid catalysts since it is strongly adsorbed on the active catalytic sites, inactivating them.16 In fact, water adsorbed on Amberlyst-16 (wet) causes a decrease in the concentration of acid sites as well as an increase in the diffusion resistance, hindering the entry of the glycerol molecule in the inner resin surface.34 These are the main factors that decrease the catalytic activity of Amberlyst-16 (wet) resulting in a lag-period in glycerol conversion in comparison to Amberlyst-15 (dry). In this manner, it is reasonable to suggest that the catalysis promoted by Amberlyst-16 (wet) occurs initially in the more accessible active sites on the outer polymer surface since few active sites of the inner resin surface are available. Glycerol diffusion resistance decreases in the course of reaction as the fluidity of the reaction medium increases with the formation of the acetins and ethanol. At this moment, it can be suggested that the active sites of the inner surface resin are now more accessible and participating in the reaction rate. Thus, as Amberlyst-16 (wet) is nearly as acid as Amberlyst-15 (dry), the results of glycerol conversion and acetin distribution in the equilibrium are similar. On the other hand, Amberlyst-15 (dry) has a greater crosslinked structure, larger pore diameter and only 1.6% of water. These features minimize the internal glycerol diffusion resistance and exclude the decrease in the concentration of active acid sites compared to Amberlyst-16 (wet) and as a result, the total glycerol consumption is achieved in 2 h only.
A similar and extensive study on the water influence on glycerol tert-butylation with isobutylene employing different macroreticular and gelular Amberlyst A-15, A-35, A-36, A-39, A-31 and A-119 acid resins in their wet and dry forms was described by Mravec and co-workers.16 Their studies showed that the highest conversion and selectivity to the desired di- and tri-ethers was obtained with all Amberlysts with macroreticular structure in dry form (A-15, A-35, A-36, A-39). Only catalysts A-15 and A-35 with very high degree of crosslinking were active and selective in dry and wet forms. The authors explained their results based mainly on the diameter of the resin pore.
Finally, the reusability and efficiency of the Amberlyst-15 (dry) resin was investigated. Thus, Amberlyst-15 (dry) was separated from the reaction mixture by filtration and reused at least thrice without significant loss of glycerol conversion as show in Figure 9. Evaporation of the residual ethyl acetate, for 24 h, at normal pressure and room temperature was sufficient to activate it.
It is worth mentioning that the ethyl acetate used in the reaction (as reagent or extraction solvent) can be salvaged for reuse while the ethanol by-product, a low toxicity substance, can be theoretically directly transformed into ethyl acetate by oxidative and dehydrogenating routes40 and fed to the process thereby increasing its efficiency.
Conclusions
We have discovered a cost-effective environmentally friendly process of producing diacetins and triacetins through acid catalyzed transesterification of glycerol with ethyl acetate, a clean alternative to the traditional methods which employ acetic anhydride and/or acetic acid. Both homogeneous (H2SO4, TsOH) and heterogeneous catalysts (Amberlyst-15TM dry, Amberlyst-16TM wet) led to an acetin mixture with 100% yield. With respect to the selectivity results, after 24 h of reaction and at 90 oC, the homogeneous catalysts showed higher selectivity toward triacetin than the Amberlyst acid resins. This result may be explained by the greater acid strength influencing the reaction rate-determining step. H2SO4 and AmberlystTM 15 dry acid resin (10% mol) proved to be the most efficient catalysts for the process since both promoted total glycerol consumption in 2.0 h at 90 ºC, with the equilibrium being established in 9 and 20 h, respectively. On the other hand, Amberlyst-16 (wet) produced similar results to Amberlyst-15 (dry) and a mixture of mono, di and triacetin with a large predominance of diacetins was obtained, however, 10 h were necessary for total glycerol consumption. A possible explanation for the longer time required is that the water adsorbed on the surface of Amberlyst-16 (wet) causes a deactivation of the acid sites and leads to an increase in the glycerol diffusion resistance, hindering the entry of the glycerol molecule in the inner resin macropore.
Supplementary Information
Supplementary data are available free of charge at http://jbcs.sbq.org.br as PDF file.
Acknowledgments
We thank CAPES and CNPq for fellowships to B. A. Meireles and V. L. P. Pereira, respectively, and FAPERJ for the financial support. We also thank Dr C. B. Barreto-Jr for graphic layout assistance.
References
1. Melero, J. A.; Iglesias, J.; Morales, G.; Green Chem. 2009, 11, 1285.
2. Demirbas A.; Energy Convers. Manage. 2009, 50, 14.
3. Sharma, Y. C.; Singh, B.; Korstad, J.; Biofuels, Bioprod. Bioref. 2011, 5, 69.
4. Guerrero-Pérez, M. O.; Rosas, J. M.; Bedia, J.; Rodríguez-Mirasol, J.; Cordero,T.; Rec. Pat. Chem. Eng. 2009, 2,11; Katryniok, B.; Kimura, H.; Skrzynska, E.; Girardon, J-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F.; Green Chem. 2011, 13, 1960.
5. Jérôme, F.; Pouilloux,Y.; Barrault, J.; ChemSusChem 2008, 1, 586.
6. Zhou, C. H.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q.; Chem. Soc. Rev. 2008, 37, 527.
7. Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F.; Green Chem. 2008, 10, 13.
8. Zheng, Y.; Chen, X.; Shen, Y.; Chem. Rev. 2008, 108, 5253.
9. Gu, Y.; Azzouzi, A.; Pouilloux, Y.; Jérôme, F.; Barrault, J.; Green Chem. 2008, 10, 164.
10. Díaz-Álvarez, A. E.; Francos, J.; Lastra-Barreira, B.; Crochet, P.; Cadierno,V.; Chem. Commun. 2011, 47, 6208.
11. Gu,Y. L.; Jerome, F.; Green Chem. 2010, 12, 1127.
12. Aggilis G.; Microbial Conversions of Raw Glycerol, Nova Science Publishers Inc.: Hauppauge, New York, 2009.
13. Rahmat, N.; Abdullah, A. Z.; Mohamed, A. R.; Renew Sustain. Energ. Rev. 2010, 14, 987; Khayoon, M. S; Hameed, B. H.; Bioresour. Technol. 2011, 102, 9229.
14. Melero, J. A.; Vicente, G.; Morales, G.; Paniagua, M.; Bustamante, J.; Fuel 2010, 89, 2011; Yang, F.; Hanna, M. A.; Sun, R.; Biotechnol. Biofuels 2012, 5, 13.
15. Di Serio, L.; Casale, R.; Santacesaria, E. T.; Energy Fuels 2010, 24, 4668.
16. Klepácová, K.; Mravec, D.; Bajus, M.; Appl. Catal., A 2005, 294, 141 and references cited therein; Garcia-Sancho, C.; Moreno-Tost, R.; Merida-Robles, J. M.; Santamaria-Gonzalez, J.; Jimenez-Lopez, A.; Torres, P. M.; Catal. Today 2011, 167, 84; Melero, J. A.; Vicente, G.; Paniagua, M.; Morales, G.; Muñoz, P.; Bioresour. Technol. 2012, 103, 142.
17. Takagaki, A.; Iwatani, K.; Nishimura, S.; Ebitani, K.; Green Chem. 2010, 12, 578.
18. Vicente, G.; Melero, J. A.; Morales, G.; Paniagua, M.; Martín, E.; Green Chem. 2010, 12, 899.
19. Silva P. H. R.; Gonçalves, V. L. C.; Mota, C. J. A.; Bioresour. Technol. 2010, 101, 6225.
20. Fukumura, T.; Toda, T.; Seki, Y.; Kubo, M.; Shibasaki-Kitakawa, N.; Yonemoto, T.; Ind. Eng. Chem. Res. 2009, 48, 1816.
21. Rezayat, M.; Ghaziaskar, H. S.; Green Chem. 2009, 11, 710.
22. Liao, X.; Zhu, Y.; Wang, S.; Chen, H.; Li, Y.; Appl. Catal., B 2010, 94, 64.
23. Casas, A.; Ruiz, J. R.; Ramos, M. J.; Pérez, A.; Energy Fuels 2010, 24, 4481.
24. Silva, L. N.; Gonçalves, V. L. C.; Mota, C. J. A.; Catal. Commun. 2010, 11, 1036.
25. Gonçalves, V. L. C.; Pinto, B. P.; Silva, J. C.; Mota, C. J. A.; Catal. Today 2008, 133-135, 673.
26. Balaraju, M.; Nikhitha, P.; Jagadeeswaraiah, K.; Srilatha, K.; Sai Prasad, P. S.; Lingaiah, N.; Fuel Process. Technol. 2010, 91, 249.
27. Ferreira, P.; Fonseca, I. M.; Ramos, A. M.; Vital, J.; Castanheiro, J. E.; Catal. Commun. 2011, 12, 573.
28. Dosuna-Rodriguez, I.; Adriany, C.; Gaigneaux, E. M.; Catal. Today 2011, 167, 56.
29. Trejda, M.; Stawicka, K.; Ziolek, M.; Appl. Catal., B 2011, 103, 404.
30. Otera, J.; Chem. Rev. 1993, 93, 1449; Otera, J.; Nishikido, J.; Sterifi cation Methods, Reactions and Applications, 2nd ed., Wiley-VCH: Weinheim, Germany, 2010.
31. Meireles, B. A.; Pereira, V. L. P.; BR PI 1.002.386-0, 2010.
32. Meireles, B. A.; Pinto, S. C.; Pereira, V. L. P.; Leitão, G. G.; J. Sep. Sci. 2011, 34, 971.
33. Morales, G.; Paniagua, M.; Melero, J. A.; Vicente, G.; Ochoa, C.; Ind. Eng. Chem. Res. 2011, 50, 5898.
34. Ertl, G.; Knozinger, H.; Schuth, F.; Weitkamp, J.; Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; Corain, B.; Zecca, M.; Jerabek, K.; J. Mol. Catal. A: Chem. 2001, 177, 3; Chang, Y.; Bae, C.; Curr. Org. Synth. 2011, 8, 208; Bozek-Winkler, E.; Gmehling, J.; Ind. Eng. Chem. Res. 2006, 45, 6648.
35. Nebel, B.; Mittelbach, M.; Uray, G.; Anal. Chem. 2008, 80, 8712.
36. Alonso, D. M.; Granados, M. L.; Mariscal, R.; Douha, A.; J. Catal. 2009, 262, 18.
37. Nakajima, K.; Hara, M.; ACS Catal. 2012, 2, 1296.
38. Ozbay, N.; Oktar, N. N.; Tapan, N. A.; Fuel 2008, 87, 1789.
39. Granollers, M.; Izquierdo, J. F.; Cunill, F.; Appl. Catal., A 2012, 435-436, 163.
40. Zonetti, P. C.; Celnik, J.; Letichevsky, S.; Gaspar, A. B.; Appel, L. G.; J. Mol. Catal. A: Chem. 2011, 334, 29; Nielsen, M.; Junge, H.; Kammer, A.; Beller, M.; Angew. Chem., Int. Ed. 2012, 5, 5711.
Submitted: June 14, 2012
Published online: February 7, 2013
Supplementary Information
The supplementary material is available in pdf: [Supplementary material]
- 1. Melero, J. A.; Iglesias, J.; Morales, G.; Green Chem 2009, 11, 1285.
- 2. Demirbas A.; Energy Convers. Manage 2009, 50, 14.
- 3. Sharma, Y. C.; Singh, B.; Korstad, J.; Biofuels, Bioprod. Bioref. 2011, 5, 69.
- 4. Guerrero-Pérez, M. O.; Rosas, J. M.; Bedia, J.; Rodríguez-Mirasol, J.; Cordero,T.; Rec. Pat. Chem. Eng. 2009, 2,11;
- Katryniok, B.; Kimura, H.; Skrzynska, E.; Girardon, J-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F.; Green Chem. 2011, 13, 1960.
- 5. Jérôme, F.; Pouilloux,Y.; Barrault, J.; ChemSusChem 2008, 1, 586.
- 6. Zhou, C. H.; Beltramini, J. N.; Fan, Y. X.; Lu, G. Q.; Chem. Soc. Rev. 2008, 37, 527.
- 7. Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F.; Green Chem. 2008, 10, 13.
- 8. Zheng, Y.; Chen, X.; Shen, Y.; Chem. Rev 2008, 108, 5253.
- 9. Gu, Y.; Azzouzi, A.; Pouilloux, Y.; Jérôme, F.; Barrault, J.; Green Chem. 2008, 10, 164.
- 10. Díaz-Álvarez, A. E.; Francos, J.; Lastra-Barreira, B.; Crochet, P.; Cadierno,V.; Chem. Commun. 2011, 47, 6208.
- 11. Gu,Y. L.; Jerome, F.; Green Chem. 2010, 12, 1127.
- 12. Aggilis G.; Microbial Conversions of Raw Glycerol, Nova Science Publishers Inc.: Hauppauge, New York, 2009.
- 13. Rahmat, N.; Abdullah, A. Z.; Mohamed, A. R.; Renew Sustain. Energ. Rev. 2010, 14, 987;
- Khayoon, M. S; Hameed, B. H.; Bioresour. Technol. 2011, 102, 9229.
- 14. Melero, J. A.; Vicente, G.; Morales, G.; Paniagua, M.; Bustamante, J.; Fuel 2010, 89, 2011;
- Yang, F.; Hanna, M. A.; Sun, R.; Biotechnol. Biofuels 2012, 5, 13.
- 15. Di Serio, L.; Casale, R.; Santacesaria, E. T.; Energy Fuels 2010, 24, 4668.
- 16. Klepácová, K.; Mravec, D.; Bajus, M.; Appl. Catal., A 2005, 294, 141 and references cited therein;
- Garcia-Sancho, C.; Moreno-Tost, R.; Merida-Robles, J. M.; Santamaria-Gonzalez, J.; Jimenez-Lopez, A.; Torres, P. M.; Catal. Today 2011, 167, 84;
- Melero, J. A.; Vicente, G.; Paniagua, M.; Morales, G.; Muñoz, P.; Bioresour. Technol. 2012, 103, 142.
- 17. Takagaki, A.; Iwatani, K.; Nishimura, S.; Ebitani, K.; Green Chem. 2010, 12, 578.
- 18. Vicente, G.; Melero, J. A.; Morales, G.; Paniagua, M.; Martín, E.; Green Chem 2010, 12, 899.
- 19. Silva P. H. R.; Gonçalves, V. L. C.; Mota, C. J. A.; Bioresour. Technol 2010, 101, 6225.
- 20. Fukumura, T.; Toda, T.; Seki, Y.; Kubo, M.; Shibasaki-Kitakawa, N.; Yonemoto, T.; Ind. Eng. Chem. Res 2009, 48, 1816.
- 21. Rezayat, M.; Ghaziaskar, H. S.; Green Chem. 2009, 11, 710.
- 22. Liao, X.; Zhu, Y.; Wang, S.; Chen, H.; Li, Y.; Appl. Catal., B 2010, 94, 64.
- 23. Casas, A.; Ruiz, J. R.; Ramos, M. J.; Pérez, A.; Energy Fuels 2010, 24, 4481.
- 24. Silva, L. N.; Gonçalves, V. L. C.; Mota, C. J. A.; Catal. Commun. 2010, 11, 1036.
- 25. Gonçalves, V. L. C.; Pinto, B. P.; Silva, J. C.; Mota, C. J. A.; Catal. Today 2008, 133-135, 673.
- 26. Balaraju, M.; Nikhitha, P.; Jagadeeswaraiah, K.; Srilatha, K.; Sai Prasad, P. S.; Lingaiah, N.; Fuel Process. Technol 2010, 91, 249.
- 27. Ferreira, P.; Fonseca, I. M.; Ramos, A. M.; Vital, J.; Castanheiro, J. E.; Catal. Commun 2011, 12, 573.
- 28. Dosuna-Rodriguez, I.; Adriany, C.; Gaigneaux, E. M.; Catal. Today 2011, 167, 56.
- 29. Trejda, M.; Stawicka, K.; Ziolek, M.; Appl. Catal., B 2011, 103, 404.
- 30. Otera, J.; Chem Rev 1993, 93, 1449;
- Otera, J.; Nishikido, J.; Sterifi cation Methods, Reactions and Applications, 2nd ed., Wiley-VCH: Weinheim, Germany, 2010.
- 31. Meireles, B. A.; Pereira, V. L. P.; BR PI 1.002.386-0, 2010
- 32. Meireles, B. A.; Pinto, S. C.; Pereira, V. L. P.; Leitão, G. G.; J. Sep. Sci. 2011, 34, 971.
- 33. Morales, G.; Paniagua, M.; Melero, J. A.; Vicente, G.; Ochoa, C.; Ind. Eng. Chem. Res. 2011, 50, 5898.
- 34. Ertl, G.; Knozinger, H.; Schuth, F.; Weitkamp, J.; Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008;
- Corain, B.; Zecca, M.; Jerabek, K.; J. Mol. Catal. A: Chem. 2001, 177, 3;
- Chang, Y.; Bae, C.; Curr. Org. Synth. 2011, 8, 208;
- Bozek-Winkler, E.; Gmehling, J.; Ind. Eng. Chem. Res. 2006, 45, 6648.
- 35. Nebel, B.; Mittelbach, M.; Uray, G.; Anal. Chem. 2008, 80, 8712.
- 36. Alonso, D. M.; Granados, M. L.; Mariscal, R.; Douha, A.; J. Catal. 2009, 262, 18.
- 37. Nakajima, K.; Hara, M.; ACS Catal. 2012, 2, 1296.
- 38. Ozbay, N.; Oktar, N. N.; Tapan, N. A.; Fuel 2008, 87, 1789.
- 39. Granollers, M.; Izquierdo, J. F.; Cunill, F.; Appl. Catal., A 2012, 435-436, 163.
- 40. Zonetti, P. C.; Celnik, J.; Letichevsky, S.; Gaspar, A. B.; Appel, L. G.; J. Mol. Catal. A: Chem. 2011, 334, 29;
- Nielsen, M.; Junge, H.; Kammer, A.; Beller, M.; Angew. Chem., Int. Ed. 2012, 5, 5711.
Publication Dates
-
Publication in this collection
28 Feb 2013 -
Date of issue
Jan 2013
History
-
Received
14 June 2012 -
Accepted
07 Feb 2013