Resumos
A síndrome de Muckle-Wells é doença autossômica dominante rara, incluída no grupo das síndromes febris hereditárias. Caracteriza-se por episódios recorrentes e autolimitados de febre, urticária, artralgia, mialgia e conjuntivite, desde a infância, relacionados com a exposição ao frio. Mais tardiamente, há perda auditiva neurossensorial progressiva. Amiloidose, a principal complicação, desenvolve-se em cerca de 25% dos casos. Associa-se a mutações no gene NLRP3 (antes CIAS1) que codifica a criopirina, proteína reguladora da produção de citocinas pró-inflamatórias, como a interleucina-1beta. Relata-se a ocorrência dessa doença incomum em quatro membros de uma única família.
Artralgia; Conjuntivite; Doenças genéticas inatas; Febre; Interleucina-1beta; Perda auditiva neurossensorial; Urticária
Muckle-Wells syndrome is a rare autosomal dominant disease that belongs to a group of hereditary febrile syndromes. It is characterized by recurrent and self-limited episodes of fever, urticaria, arthralgia, myalgia and conjunctivitis since childhood, which are related to exposure to cold temperatures. Lately, progressive sensorineural hearing loss occurs. Amyloidosis is the main complication and can be found in about 25% of the cases. It has been demonstrated that there is an association with mutations in the NLRP3 gene, which codifies cryopyrin, a protein responsible for regulating the production of proinflammatory cytokines, such as interleukin-1Beta. The authors report four cases of the disease within a family.
Arthralgia; Conjunctivitis; Fever; Genetic diseases, inborn; Hearing loss, sensorineural; Interleukin-1Beta; Urticaria
CASO CLÍNICO
Síndrome de Muckle-Wells em quatro membros de uma família* * Trabalho realizado no Serviço de Dermatologia do Hospital das Clínicas da Universidade Federal de Minas Gerais (UFMG) - Belo Horizonte (MG), Brasil.
Ana Francisca Junqueira Ribeiro PereiraI; Luciana Baptista PereiraII; Everton Carlos Siviero do ValeIII; Leandro Augusto TanureIV
IMédica residente do Serviço de Dermatologia do Hospital das Clínicas da Universidade Federal de Minas Gerais (UFMG) - Belo Horizonte (MG), Brasil
IIProfessora Assistente do Serviço de Dermatologia da Faculdade de Medicina da Universidade Federal de Minas Gerais (UFMG) - Belo Horizonte (MG), Brasil
IIIProfessor Assistente do Serviço de Dermatologia do Hospital das Clínicas da Universidade Federal de Minas Gerais (UFMG) - Belo Horizonte (MG), Brasil
IVMédico pós-graduando do Serviço de Reumatologia do Hospital das Clínicas da Universidade Federal de Minas Gerais (UFMG) - Belo Horizonte (MG), Brasil
Endereço para correspondência Endereço para correspondência: Ana Francisca Junqueira Ribeiro Pereira Rua Ouro Preto, 1410/302. Santo Agostinho 30170 040 - Belo Horizonte - MG Tel./Fax: 31 3292 7773 / Fax: 31 3225 3110 email: anafranciscajunqueira@hotmail.com
RESUMO
A síndrome de Muckle-Wells é doença autossômica dominante rara, incluída no grupo das síndromes febris hereditárias. Caracteriza-se por episódios recorrentes e autolimitados de febre, urticária, artralgia, mialgia e conjuntivite, desde a infância, relacionados com a exposição ao frio. Mais tardiamente, há perda auditiva neurossensorial progressiva. Amiloidose, a principal complicação, desenvolve-se em cerca de 25% dos casos. Associa-se a mutações no gene NLRP3 (antes CIAS1) que codifica a criopirina, proteína reguladora da produção de citocinas pró-inflamatórias, como a interleucina-1beta. Relata-se a ocorrência dessa doença incomum em quatro membros de uma única família.
Palavras-chave: Artralgia; Conjuntivite; Doenças genéticas inatas; Febre; Interleucina-1beta; Perda auditiva neurossensorial; Urticária
INTRODUÇÃO
A síndrome de Muckle-Wells (MWS, Muckle-Wells syndrome) é doença autossômica dominante rara, pertencente ao grupo das doenças autoinflamatórias hereditárias. Associa-se a mutações no gene NLRP3 (antes conhecido como CIAS1) que codifica a criopirina. Essa proteína é responsável por regular a produção de citocinas pró-inflamatórias, como a interleucina-1beta. Já foram descritas mais de 50 mutações neste gene, algumas relacionadas com diferentes fenótipos. O motivo da apresentação dos casos se deve à raridade da síndrome.
RELATO DOS CASOS
caso 1
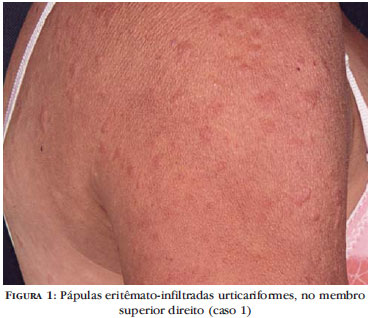
Paciente do sexo feminino, de 43 anos, apresenta quadro recorrente de urticária (Figuras 1, 2 e 3), artralgias e edema de joelhos e tornozelos, além de hiperemia conjuntival, desde os primeiros meses de vida. Os episódios duram cerca de dois a três dias e se relacionam com a exposição ao frio. O quadro cutâneo costuma ser pouco pruriginoso, havendo discreta ardência local. Queixa-se de hipoacusia há alguns anos, tendo sido observada perda auditiva neurossensorial à audiometria. O fator antinúcleo (FAN) é persistentemente negativo, bem como crioglobulinas, anticardiolipina e anticoagulante lúpico. A velocidade de hemossedimentação (VHS) e a proteína C reativa (PCR) são elevadas as dosagens séricas de C3 e C4 são normais. Revisão laboratorial, exame de urina-rotina e radiografia do tórax e das mãos não apresentaram alterações. Exame histopatológico de lesão urticariforme de pele evidenciou, na derme, discreto infiltrado inflamatório perivascular linfo-histiocitário, com alguns neutrófilos e eosinófilos, além de edema e agressão inflamatória vascular, sugerindo vasculite neutrofílica de pequenos vasos (Figura 4). A imunofluorescência direta foi negativa para IgA, IgG, IgM e C3.
caso 2
Paciente do sexo feminino, de 24 anos, com episódios de dor e edema de tornozelos, associados à urticária não-pruriginosa, relacionados ao frio, desde a infância. Exames laboratoriais mostraram VHS elevada, FAN negativo, dosagens de C3 e C4 normais e exame de urina-rotina sem alterações. A biópsia de pele revelou discreto infiltrado inflamatório perivascular linfocitário, com poucos neutrófilos, na derme superior, com agressão aos vasos. A imunofluorescência direta foi negativa. Audiometria evidenciou discreta perda auditiva neurossensorial à esquerda.
caso 3
Paciente do sexo masculino, de 20 anos, apresenta surtos de urticária, hiperemia conjuntival (Figura 5) e artralgia nos joelhos, desde a primeira infância, que pioram com a exposição ao frio. As lesões cutâneas não são pruriginosas, ocorrendo apenas sensação de aumento da temperatura local. Há surdez neurossensorial à esquerda. A VHS e a PCR são elevadas, o FAN é negativo e dosagens de C3 e C4 são normais. Ao exame de urina rotina, diagnosticou-se hipocalciúria. Exame histopatológico de pele mostrou apenas dermite crônica. Estudo genético evidenciou cariótipo normal e a pesquisa de quebras cromossômicas foi negativa.
caso 4
Paciente do sexo feminino, de 17 anos, com quadro recalcitrante de urticária não pruriginosa (Figura 6) e dor nos tornozelos, desde os primeiros anos de vida, desencadeado por exposição ao frio. Há discreta perda auditiva neurossensorial bilateral. O FAN é negativo e exame de urina-rotina, VHS, PCR e dosagem de C3 e C4 são normais. À histopatologia, observou-se discreto exsudato linfocitário, com poucos neutrófilos, ao redor de pequenos vasos, na derme superior, havendo agressão vascular, além de epiderme no limite da normalidade. A imunofluorescência direta foi negativa.
DISCUSSÃO
A MWS caracteriza-se por episódios recorrentes e autolimitados de febre, urticária, artralgia, mialgia e conjuntivite, desde a infância, eventualmente relacionados com a exposição ao frio.1,2 Cefaleia,3 dor abdominal3,4 e retardo do crescimento4 podem ainda fazer parte do quadro clínico. Mais tardiamente, há perda auditiva neurossensorial progressiva.1 Amiloidose tipo AA, presente em cerca de 25% dos casos, é a principal complicação da doença, com acometimento predominantemente renal.1,2
Geralmente, a erupção cutânea é a primeira manifestação a ser notada, caracterizando-se por lesões maculopapulares urticariformes, de caráter migratório.5 O prurido é incomum, podendo haver sensação de queimação.5 Os achados histológicos, de infiltrado inflamatório dérmico perivascular, composto predominantemente por polimorfonucleares, contrasta com o infiltrado típico de eosinófilos e linfócitos da urticária clássica, levando alguns autores a usar o termo pseudo-urticária, para designar a erupção da MWS.5 Apesar das manifestações dermatológicas mais características serem as lesões urticariformes, El-Darouti et al. descreveram seis casos de lesões esclerodermoides, com hiperpigmentação, esclerose e hipertricose.4
Além da MWS, a síndrome autoinflamatória familiar, relacionada ao frio (FCAS, familial cold autoinflammatory syndrome), e a síndrome neurocutâneo-articular crônica infantil (CINCA, chronic infantile neurological cutaneous articular syndrome) também são doenças febris hereditárias, de padrão autossômico dominante. Igualmente, as três entidades associam-se a mutações no gene NLRP3 (antes CIAS1), localizado no cromossomo 1.6 Maksimovic et al. observaram fenótipos das três síndromes em uma mesma família7 e Hoffman et al. encontraram diferentes fenótipos intrafamiliares, como resultado de uma mesma mutação no gene NLRP3.8 Já Dodé et al. identificaram a mesma mutação em duas famílias com síndrome de Muckle-Wells e em outras duas com FCAS, não havendo origem étnica semelhante entre elas.9
Todos esses achados sugerem que a síndrome de MWS, a FCAS e a CINCA constituam diferentes expressões fenotípicas de uma mesma doença,3,7,10,11 havendo casos descritos de sobreposição dessas síndromes. Acredita-se que haja correlação entre genótipo e fenótipo, levando a características clínicas, penetrância e gravidade distintas.12 Além disso, fatores ambientais e genes modificadores podem determinar diferentes fenótipos.3,6 A FCAS parece representar o fenótipo mais brando, ao passo que a CINCA ocupa o pólo mais grave. Enquanto a FCAS apresenta-se basicamente com urticária ao frio, febre e artralgia, na MWS desenvolvem-se ainda perda auditiva e amiloidose. Já na CINCA, há alterações do sistema nervoso central e artropatia, sendo o surgimento da urticária muito precoce, ainda no período neonatal. As manifestações neurológicas, por conta da meningite crônica asséptica, ocorrem em quase todos os pacientes com CINCA e variam desde cefaleia crônica, vômitos e papiledema até plegia espástica e epilepsia.5
Em acordo com os diversos estudos de famílias com portadores de MWS, os casos ora relatados respeitam o padrão autossômico dominante de ocorrência. Entre os quatro membros desta família, podem-se notar graus variáveis de sinais e sintomas, analogamente ao que se descreve na literatura. Desse modo, seria possível classificar os pacientes estudados como portadores de uma das síndromes (FCAS, MWS ou CINCA), isto é, portadores de diferentes espectros de uma só doença. Porém, todos os quatro pacientes apresentam, ainda que em graus variados, perda auditiva neurossensorial, manifestação presente na MWS. Além disso, nenhum deles apresentou até o momento sintomas neurológicos ou artrite franca, o que enfraquece a hipótese de algum destes pacientes ser portador da CINCA.
As síndromes autoinflamatórias são um grupo de doenças inflamatórias sistêmicas, que não são causadas por nenhum patógeno ou autoanticorpo. Elas resultam da regulação aberrante das vias de sinalização de citocinas, levando à inflamação persistente ou descontrolada.11 Ainda não se sabe se essas doenças poderiam originar-se de respostas normais a alguma infecção, porém é, justamente, o estado inflamatório persistente na ausência de infecção aparente o ponto característico desta classe de doenças.11
Síndromes febris periódicas, acompanhadas de erupção urticariforme, tais como: a FCAS, a MWS e a CINCA, foram associadas a mutações no gene NLRP3 (antes CIAS1) e têm sido conjuntamente designadas síndromes periódicas, associadas à criopirina (CAPS, cryopyrin associated periodic syndromes ).10 Esse grupo é, atualmente, considerado um outro espectro das principais síndromes autoinflamatórias.10 Por consequência, o conceito de síndrome autoinflamatória tem sido ampliado, a fim de incluir outras doenças hereditárias raras com ou sem febre periódica, como pioderma gangrenoso e artrite estéril piogênica.10 Em contrapartida, a presença de algumas dessas mutações genéticas em enfermidades menos incomuns, como doença de Behçet e doença de Crohn, levou à expansão do espectro das síndromes autoinflamatórias para as também chamadas doenças colagenose-símiles.10 Todas elas compartilham a mesma fisiopatologia, com hiperativação de neutrófilos, monócitos ou macrófagos, além de predisposição genética à desregulação da sinalização da imunidade inata.10 Avanços no entendimento da base genética das síndromes periódicas, associadas à criopirina, propiciaram a descoberta de moléculas de sinalização de citocinas, que são essenciais para a regulação de certas vias inflamatórias.11 A criopirina é responsável pela via de ativação da interleucina-1, atuando como parte de um complexo macromolecular maior, chamado inflamassomo NALP3.11 Este, por sua vez, ativa a interleucina-1beta e a interleucina-18, o que resulta em ativação imunológica, inflamação e injúria tecidual.11 A criopirina também regula a via de ativação do fator nuclear kappa B (NFkB) e a apoptose.3 Entretanto, o mecanismo, pelo qual essas vias são aberrantemente reguladas nas síndromes autoinflamatórias hereditárias, permanece obscuro, apesar de mutações, no gene NLRP3 (antes CIAS1), parecerem causar uma hiper-regulação persistente em tais vias,11 acarretando um intenso efeito pró-inflamatório6. Em contrapartida, a heterogeneidade genética das criopirinopatias tem sido recentemente ressaltada.6,13 Neven et al. conseguiram identificar mutações no gene NLRP3, em apenas 60% dos pacientes analisados.13
As opções terapêuticas são: os anti-inflamatórios, anti-histamínicos,1 colchicina e corticosteroides, todos com resposta insatisfatória. Ultimamente, tem sido empregado um homólogo recombinante da interleucina-1beta (anakinra), com bons resultados, observando-se, até mesmo, a melhora da hipoacusia e da amiloidose2, quando introduzido precocemente.
A resposta dramática das síndromes febris hereditárias a bloqueadores das vias de ativação de citocinas, como o anakinra, corroborando o papel crucial das citocinas inflamatórias, por exemplo a interleucina-1beta, na patogênese dessas síndromes.3,11,12 Em vista disso, estas citocinas passaram a ser potenciais alvos terapêuticos para esse grupo de doenças. Melhor elucidação desses mecanismos poderá, no futuro, facilitar o desenvolvimento de estratégias biológicas de tratamento, assegurando uma intervenção imunossupressora altamente eficaz e seletiva.
Recebido em 18.08.2008.
Aprovado pelo Conselho Consultivo e aceito para publicação em 15.10.2008.
Conflito de interesse: Nenhum
None Suporte financeiro: Nenhum
- 1. Haas N, Küster W, Zuberbier T, Henz BM. Muckle-Wells syndrome: clinical and histological skin findings compatible with cold air urticaria in a large kindred. Br J Dermatol. 2004;151:99-104.
- 2. Kagami S, Saeki H, Kuwano Y, Imakado S, Tamaki K. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-21.
- 3. Farasat S, Aksentijevich I, Toro JR. Autoinflammatory diseases: clinical and genetic advances. Arch Dermatol. 2008;144:392-402.
- 4. El-Darouti MA, Marzouk SA, Abdel-Halim MR. Muckle-Wells syndrome: report of six cases with hyperpigmented sclerodermoid skin lesions. Int J Dermatol. 2006;45:239-44.
- 5. Neven B, Prieur AM, Quartier dit Maire P; Medscape. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-9.
- 6. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID). Arthritis Rheum. 2002;46:3340-8.
- 7. Maksimovic L, Stirnemann J, Caux F, Ravet N, Rouaghe S, Cuisset L, et al. New CIAS1 mutation and anakinra efficacy in overlapping of Muckle-Wells and familial cold autoinflammatory syndromes. Rheumatology. 2008;47:309-10.
- 8. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301-5.
- 9. Dodé C, Le Dû N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002;70:1498-506.
- 10. Kanasawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-18.
- 11. Shinkai K, McCalmont TH, Leslie KS. Cryopyrinassociated periodic syndromes and autoinflammation. Clin Exp Dermatol. 2007;33:1-9.
- 12. Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607-12.
- 13. Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, et al. Molecular basis of the espectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS and FCU. Blood. 2004;103:2809-15.
Datas de Publicação
-
Publicação nesta coleção
27 Jan 2011 -
Data do Fascículo
Dez 2010
Histórico
-
Recebido
18 Ago 2008 -
Aceito
15 Out 2008