Resumos
INTRODUÇÃO: A Síndrome de Dandy Walker consiste em uma anormalidade congênita do sistema nervoso central, caracterizada por deficiência do desenvolvimento das estruturas médias cerebelares, dilatação cística de fossa posterior comunicando- se com o quarto ventrículo e deslocamento ascendente dos seios transversais, tentório e tórcula. Entre os sinais clínicos estão protuberância occipital, aumento progressivo do crânio, arqueamento das fontanelas anteriores, papiledema, ataxia, distúrbios da marcha, nistagmo e comprometimento intelectual. OBJETIVOS: Descrever um caso de paciente feminino, 13 anos com diagnóstico desta síndrome e perda auditiva bilateral submetida à cirurgia de implante coclear sob anestesia local e sedação. RELATO DE CASO: CGS, 13 anos, sexo feminino, foi referida ao Serviço de Otorrinolaringologia do Instituto paranaense de Otorrinolaringologia com diagnóstico de "síndrome de Dandy-Walker" para avaliação otorrinolaringológica por perda auditiva bilateral sem resposta ao uso de aparelhos de amplificação sonora. COMENTÁRIOS FINAIS: O campo da cirurgia de implante coclear está crescendo rapidamente. Acreditamos que a presença da síndrome de Dandy-Walker não pode ser considerada uma contra indicação para a realização da cirurgia de implante coclear, sendo que não houve intercorrências cirúrgicas devido às alterações neurológicas com resultados muito favoráveis para a paciente que apresenta excelente discriminação. Apresenta menor necessidade de leitura labial com melhora na qualidade da fala.
perda auditiva; implante coclear; síndrome de Dandy-Walker
INTRODUCTION: Dandy Walker Syndrome is a congenital abnormality in the central nervous system, characterized by a deficiency in the development of middle cerebelar structures, cystic dilatation of the posterior pit communicating with the fourth ventricle and upward shift of the transverse sinuses, tentorium and dyes. Among the clinical signs are occipital protuberances, a progressive increase of the skull, bowing before the fontanels, papilledema, ataxia, gait disturbances, nystagmus, and intellectual impairment. OBJECTIVES: To describe a case of female patient, 13 years old with a diagnosis of this syndrome and bilateral hearing loss underwent cochlear implant surgery under local anesthesia and sedation. CASE REPORT: CGS, 13 years old female was referred to the Otolaryngological Department of Otolaryngology Institute of Parana with a diagnosis of "Dandy-Walker syndrome" for Otolaryngological evaluation for bilateral hearing loss with no response to the use of hearing aids. FINAL COMMENTS: The field of cochlear implants is growing rapidly. We believe that the presence of Dandy-Walker syndrome cannot be considered a contraindication to the performance of cochlear implant surgery, and there were no surgical complications due to neurological disorders with very favorable results for the patient who exhibits excellent discrimination. It has less need for lip reading with improvement in speech quality.
hearing loss; cochlear implants; Dandy-Walker syndrome
RELATO DE CASO
Implante coclear em paciente com síndrome de Dandy-walker
Adriana Kosma Pires de OliveiraI; Rogerio HamerschmidtII; Marcos MocelinIII; Rodrigo K. RezendeIV
IMédica Otorrinolaringologista do Hospital Paranaense de Otorrinolaringologia
IIProfessor da Disciplina de Otorrinolaringologia da Universidade Federal do Paraná. Médico Otorrinolaringologista do Hospital Paranaense de Otorrinolaringologia
IIIChefe do Serviço de Otorrinolaringologia do Hospital de Clinicas da Universidade Federal do Paraná. Chefe do Serviço de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná
IVMédico Residente de Otorrinolaringologia no Hospital de Clínicas da Universidade Federal do Paraná
Endereço para correspondência Endereço para correspondência: Adriana Kosma Pires de Oliveira Rua Edmundo de Amicis, 316 Curitiba / PR - Brasil - CEP: 81810-160 E-mail: adri_pires@hotmail.com
RESUMO
INTRODUÇÃO: A Síndrome de Dandy Walker consiste em uma anormalidade congênita do sistema nervoso central, caracterizada por deficiência do desenvolvimento das estruturas médias cerebelares, dilatação cística de fossa posterior comunicando- se com o quarto ventrículo e deslocamento ascendente dos seios transversais, tentório e tórcula. Entre os sinais clínicos estão protuberância occipital, aumento progressivo do crânio, arqueamento das fontanelas anteriores, papiledema, ataxia, distúrbios da marcha, nistagmo e comprometimento intelectual.
OBJETIVOS: Descrever um caso de paciente feminino, 13 anos com diagnóstico desta síndrome e perda auditiva bilateral submetida à cirurgia de implante coclear sob anestesia local e sedação.
RELATO DE CASO: CGS, 13 anos, sexo feminino, foi referida ao Serviço de Otorrinolaringologia do Instituto paranaense de Otorrinolaringologia com diagnóstico de "síndrome de Dandy-Walker" para avaliação otorrinolaringológica por perda auditiva bilateral sem resposta ao uso de aparelhos de amplificação sonora.
COMENTÁRIOS FINAIS: O campo da cirurgia de implante coclear está crescendo rapidamente. Acreditamos que a presença da síndrome de Dandy-Walker não pode ser considerada uma contra indicação para a realização da cirurgia de implante coclear, sendo que não houve intercorrências cirúrgicas devido às alterações neurológicas com resultados muito favoráveis para a paciente que apresenta excelente discriminação. Apresenta menor necessidade de leitura labial com melhora na qualidade da fala.
Palavras-chave: perda auditiva, implante coclear, síndrome de Dandy-Walker.
INTRODUÇÃO
O relato clássico, feito por Dandy & Blackfan em 1914*, revelou casos em autópsias com hidrocefalia severa supratentorial, dilatação cística do quarto ventrículo, vermis pequeno, afastamento dos hemisférios cerebelares, ausência do teto do quarto ventrículo, espessamento e opacificação da pia-aracnoide das cisternas da base do crânio e dilatação do aqueduto (1).
A síndrome de Dandy-Walker (SDW) é uma síndrome não familiar, caracterizada por dilatação cística do quarto ventrículo e por aplasia ou hipotrofia parcial ou total do vermis cerebelar. Geralmente apresenta atresia dos forames de Lushka e Magendie. Em 75% dos casos ocorrem outras malformações cerebrais como agenesia do corpo caloso, heteropsias, lissencefalia, estenose do aqueduto de Sylvius (2-3). Gardner et al** propuseram que a SDW, juntamente com outras síndromes (Arnold-Chiari, cisto aracnoide de cerebelo e siringomielia), seriam manifestações de uma mesma doença (1).
Alguns estudos mostram uma incidência de aproximadamente 70% de relação entre a SDW e anomalias sistêmicas (1). Pouco se sabe sobre malformações congênitas das estruturas da fossa posterior, suas alterações genéticas foram mapeadas para o cromossomo 3q (4-5), mas o gene ao certo não localizado, porém sabe-se que a base do processo de desenvolvimento das estruturas da fossa posterior é a natureza para as malformações cerebelares humanas (5). Sabe-se também que as estruturas cerebelares se desenvolvem precocemente no período embrionário até os primeiros anos pós-natais, este acontecimento deixaria o cerebelo vulnerável a um largo espectro de desordens do seu desenvolvimento (6).
A patogenia desta síndrome é controversa, porém a teoria mais aceita é a de que a folha do desenvolvimento dos forames de Lushka e Magendie, durante o quarto mês de vida fetal, leva ao abaulamento cístico do quarto ventrículo. Novas teorias propuseram que a SDW decorreria de uma falha no desenvolvimento no teto do rombencéfalo, tendo este como causa um efeito teratogênico (1). Alguns trabalhos sugerem que o uso de warfarin em longo prazo seria responsável pelo desenvolvimento da SDW em 1-2% dos fetos expostos (7). O que vem reforçar ainda mais esta teoria é que um estudo realizado por Hart et al., mostrou que não há relação entre grau de hidrocefalia e o tamanho do cisto da fossa posterior, atenuação do vermis ou permeabilidade do quarto ventrículo e em alguns casos a hidrocefalia estava ausente (1).
A SDW é uma entidade heterogênica de hipoplasia de vermis cerebelar, sendo recentemente identificado um gene associado à ligação com X-HPRT (8), também relacionado com doença de gânglios da base.
Clinicamente pode haver moderado atraso do desenvolvimento psicomotor, microcefalia, hipotonia, mas a sintomatologia predominante se refere à hidrocefalia, geralmente nos dois primeiros anos de vida, esta porém, pode ser ignorada, aparecendo tardiamente (primeira ou segunda década de vida) (2-9). A hidrocefalia se dá pela obstrução dos forames de Lushka e Mangedie (1). Algumas alterações oculares são descritas na SDW, como: coloboma corioretiniano (10), nistagmo (11-13). Pode haver retardo mental (50%), espasticidade (ao invés de hipotonia), convulsões, vômitos, tudo dependendo do grau da malformação cerebelar (3). Em pacientes com vermis com duas fissuras e conformações praticamente normais, as funções cerebrais são também praticamente normais sem associações com outras malformações. Já em pacientes com severas malformações do cerebelo, vermis com apenas uma ou nenhuma fissura, é comum o retardo mental severo e outras malformações do sistema nervoso central, como agenesia de corpo caloso. Divide-se, assim, a SDW em dois grandes grupos conforme as malformações anteriores para a determinação do prognóstico intelectual (12-13). São relatados na literatura casos de coexistência de grandes hemangiomas cutâneos faciais com a SDW (14). Outras síndromes onde coexistem malformações cerebrais e oculares são relatadas como síndrome de Neuhauser (MMMM - Megalocornea, macrocephaly, mental and motor retardation), onde são encontrados atrofia cortical, aumento do quarto ventrículo, hipoplasia do corpo caloso. Tudo isto considerado uma variante da SDW, juntamente com megalocórnea (15). Para o diagnóstico há necessidade da ressonância magnética com imagens de boa qualidade da vista axial do vermis cerebelar e imagens em T2 (12). Os achados neuroradiológicos são característicos, como a dilatação cística do quarto ventrículo e as alterações no vermis cerebelar (2), além de outras já citadas. A SDW deve sempre ser acompanhada por uma equipe multidisciplinar. O tratamento da hidrocefalia sempre será cirúrgico, através de uma derivação ventrículo-peritoneal (2).
A surdez neurosensorial bilateral pode fazer partedo quadro clinico da síndrome sendo que descrevemos um caso cuja paciente foi submetida a implante cocler por apresentar surdez profunda bilateral.
O objetivo deste artigo é descrever um caso de paciente feminino, 13 anos com diagnóstico desta síndrome e perda auditiva bilateral submetida à cirurgia de implante coclear sob anestesia local e sedação.
RELATO DE CASO
CGS, 13 anos, sexo feminino, foi referida ao Serviço de Otorrinolaringologia do Instituto paranaense de Otorrinolaringologia com diagnóstico de "síndrome de Dandy-Walker" para avaliação otorrinolaringológica por perda auditiva bilateral sem resposta ao uso de aparelhos de amplificação sonora.
A paciente em questão realiza acompanhamento com a neurologia desde nascimento, com história de rubéola gestacional, catarata em ambos os olhos e cirurgia neurológica prévia por hidrocefalia, não existindo consanguinidade entre os pais. Criança com exame de imagem compatível com síndrome de Dandy-Walker, com coleção na fossa posterior e com comunicação com o quarto ventrículo, associada com sinais de hipoplasia dos hemisférios cerebelares e do vermis. Cariótipo não realizado. Déficit neuro- psicomotor leve.
A criança preencheu critérios para implante coclear. Foi submetida por avaliação médica otorrinolaringológica, neurológica, fonoaudiológica e pscicológica.
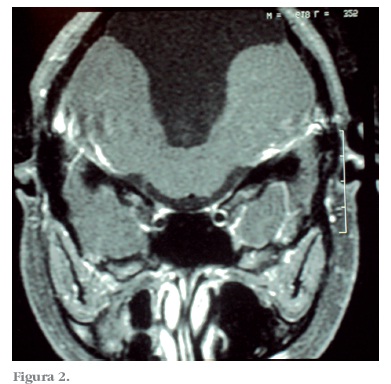
Na audiometria apresentava perda auditiva sensório neural profunda bilateral. Na avaliação eletrofisiológica da audição não foi possível detectar potenciais evocados de tronco cerebral, bilateralmente. Na emissão otoacústica ausência de resposta bilateralmente A ressonância magnética de encéfalo mostra hipoplasia de verme cerebelar associado à mega cisterna magna com comunicação com o quarto ventrículo (Figuras 1 e 2).
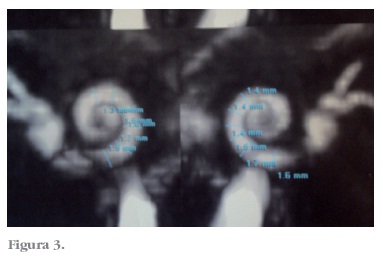
A ressonância de ouvidos as configurações dos ouvidos internos são simétricas e de configuração normal. Não há realces anômalos relacionados às mesmas ou ao restante do encéfalo. Os espaços líquidos da cóclea tem espessura normal, variando de 1,3 a 1,7 mm a direita e de 1,4 a 1,7 mm à esquerda. Os nervos cocleares foram identificados e tem espessura normal de 0,6 mm (Figura 3).
Foi submetida a implante coclear de ouvido direito. A prótese utilizada foi um implante nucleus freedom do Cochlear Comparation®. A cirurgia foi realizada sob anestesia local e sedação por acesso retroauricular, confecção do nicho da unidade externa, mastoidectomia fechada, timpanotomia posterior, cocleostomia, inserção do componente interno e telemetria. Todos os passos realizados sem intercorrências. O implante foi ativado 1 mês após a cirurgia com ótima percepção sonora no momento da ativação. A paciente apresenta excelentes níveis de discriminação com menor necessidade de leitura labial e excelente resposta a fonoterapia com espetacular melhorai da qualidade da fala.
Sem dúvida os resultados obtidos trazem repercussão na qualidade de vida da paciente além de proporcionar melhor integração social.
COMENTÁRIOS FINAIS
O campo da cirurgia de implante coclear está crescendo rapidamente, devido à melhora na qualidade dos implantes, cirurgias menos invasivas, e maior divulgação deste tipo de tratamento da surdez. Os implantes cocleares são próteses extremamente caras que parcialmente substituem as funções da cóclea (20). A cirurgia atualmente é muito mais rápida e menos invasiva que há anos atrás, com menores incisões e menor morbidade para o paciente.
A paciente embora portadora da síndrome de Dandy -Walker não apresenta déficit significativo no desenvolvimento neuropscicomotor, preenchendo os critérios já estabelecidos na literatura para realização da cirurgia de implante coclear. Foi avaliada por equipe multidisciplinar sem contra indicações para a realização do procedimento. Já existem casos de pacientes com mesma síndrome submetidos à cirurgia de implante coclear se, intercorrências descritos na literatura (21) e com bons resultados apesar das comorbidades.
Em nosso serviço optamos por realizar esta cirurgia sob anestesia local e sedação. Esta forma de anestesia traz uma morbidade cirúrgico/anestésica menor alem de uma recuperação pós operatória mais rápida e menores custos hospitalares quando comparada a anestesia geral e houve perfeita colaboração por parte da paciente.
Acreditamos que a presença da síndrome de Dandy-Walker não pode ser considerada uma contra indicação para a realização da cirurgia de implante coclear, sendo que não houve intercorrências cirúrgicas devido às alterações neurológicas com resultados muito favoráveis para a paciente que apresenta excelente discriminação. Apresenta menor necessidade de leitura labial com melhora na qualidade da fala.
Artigo recebido em 2 de Junho de 2010.
Artigo aprovado em 1º de Agosto de 2010.
Instituição: Hospital paranaense de otorrinolaringologia. Curitiba / PR - Brasil.
- 1. Diament A. Neurologia infantil 3a ed. São Paulo: Atheneu; 1996.
- 2. Rosenberg S. Neuropediatria. São Paulo: Sarvier; 1995.
- 3. Yilmaz MA. The site on Dandy Walker Syndrome [homepage on the Internet]. [cited 2005 Aug 29]. Available from: http://www.geocities.com/murat_yil/dandy.html
- 4. Parisi MA, Dobyns WB. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab, 2003; 80(1-2):36-53.
- 5. Chizhikov V, Millen KJ. Development and malformations of the cerebellum in mice. Mol Genet Metab. 2003, 80(1-2):54-65.
- 6. Ten Donkelaar HJ, Lammens M, Wesseling P, Thijssen HO, Renier WO. Development and developmental disorders of the human cerebellum. J Neurol. 2003, 250(9):1025-36.
- 7. Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med. 1998, 338(16): 1128-37.
- 8. Daufenbach DR, Ruttum MS, Pulido JS, Keech RV. Chorioretinal colobomas in a pediatric population. Ophthalmology. 1998, 105(8):1455-8. Comment in: Ophthalmology. 1999, 106(4):645-6.
- 9. Niesen CE. Malformations of the posterior fossa: current perspectives. Semin Pediatr Neurol. 2002, 9(4):320-34.
- 10. Coats DK, Paysse EA, Levy ML. PHACE: a neurocutaneous syndrome with important ophthalmologic implications: case report and literature review. Ophthalmology. 1999, 106(9):1739-41.
- 11. Kumandas S, Akcakus M, Coskun A, Gumus H. Joubert syndrome: review and report of seven new cases. Eur J Neurol. 2004, 11(8):505-10.
- 12. Klein O, Pierre-Kahn A, Boddaert N, Parisot D, Brunelle F. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst. 2003, 19(78):484-9.
- 13. Boddaert N, Klein O, Ferguson N, Sonigo P, Parisot D, Hertz-Pannier L, et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology. 2003, 45(5):320-4.
- 14. Reese V, , Paller AS, Esterly NB, Ferriero D, Levy ML, et al. Association of facial hemangiomas with Dandy-Walker and other posterior fossa malformations. J Pediatr 1993, 122(3):379-84.
- 15. Balci S, Teksam O, Gedik S. Megalocornea, macrocephaly, mental and motor retardation: MMMM syndrome (Neuhauser syndrome) in two sisters with hypoplastic corpus callosum. Turk J Pediatr. 2002, 44(3):274-7.
- 16. Pagon RA, Clarren SK, Milam DF Jr, Hendrickson AE. Autosomal recessive eye and brain anomalies: Warburg syndrome. J Pediatr. 1983, 102(4):542-6.
- 17. Golden JA. Cell migration and cerebral cortical development. Neuropathol Appl Neurobiol. 2001, 27(1):22-8.
- 18. Ruibal Francisco JL, Sánchez Buron P, Piñero Martinez E, Bueno Lozano G. [Turner's syndrome. Relationship between the karyotypes and malformations and associated diseases in 23 patients]. An Esp Pediatr. 1997, 47(2):167-71. Spanish.
- 19. Kawaguchi T, Jokura H, Kusaka Y, Shirane R, Yoshimoto T. Intraoperative direct neuroendoscopic observation of the aqueduct in Dandy-Walker malformation. Acta Neurochir (Wien). 2003, 145(1):63-7
- 20. Chakrabarty A, Tarneja VK, Singh VK. Cochlear implant: anesthesia challenges. MJAFI Hournal, 1994, 60:351-6.
- 21. Sharon LC*, Laurie Mb, Evan J, Alok S, Tracy S. Successful cochlear implantation in a child with Keratosis, Icthiosis and Deafness (KID) Syndromeand Dandy-Walker malformation. International Journal of Pediatric Otorhinolaryngology. 2008;72,693-698.
Endereço para correspondência:
Datas de Publicação
-
Publicação nesta coleção
15 Ago 2012 -
Data do Fascículo
Set 2012
Histórico
-
Recebido
02 Jun 2010 -
Aceito
01 Ago 2010