Abstract
We investigated whether hepatic artery endothelium may be the earliest site of injury consequent to liver ischemia and reperfusion. Twenty-four heartworm-free mongrel dogs of either sex exposed to liver ischemia/reperfusion in vivo were randomized into four experimental groups (N = 6): a) control, sham-operated dogs, b) dogs subjected to 60 min of ischemia, c) dogs subjected to 30 min of ischemia and 60 min of reperfusion, and d) animals subjected to 45 min of ischemia and 120 min of reperfusion. The nitric oxide endothelium-dependent relaxation of hepatic artery rings contracted with prostaglandin F2a and exposed to increasing concentrations of acetylcholine, calcium ionophore A23187, sodium fluoride, phospholipase-C, poly-L-arginine, isoproterenol, and sodium nitroprusside was evaluated in organ-chamber experiments. Lipid peroxidation was estimated by malondialdehyde activity in liver tissue samples and by blood lactic dehydrogenase (LDH), serum aspartate aminotransferase (AST) and serum alanine aminotransferase (ALT) activities. No changes were observed in hepatic artery relaxation for any agonist tested. The group subjected to 45 min of ischemia and 120 min of reperfusion presented marked increases of serum aminotransferases (ALT = 2989 ± 1056 U/L and AST = 1268 ± 371 U/L; P < 0.01), LDH = 2887 ± 1213 IU/L; P < 0.01) and malondialdehyde in liver samples (0.360 ± 0.020 nmol/mgPT; P < 0.05). Under the experimental conditions utilized, no abnormal changes in hepatic arterial vasoreactivity were observed: endothelium-dependent and independent hepatic artery vasodilation were not impaired in this canine model of ischemia/reperfusion injury. In contrast to other vital organs and in the ischemia/reperfusion injury environment, dysfunction of the main artery endothelium is not the first site of reperfusion injury.
Endothelium; Hepatic artery; Nitric oxide; Liver; Ischemia/reperfusion injury
Braz J Med Biol Res, June 2007, Volume 40(6) 857-865
Endothelium-dependent and -independent hepatic artery vasodilatation is not impaired in a canine model of liver ischemia-reperfusion injury
L.C. Miranda, F. Viaro, R. Ceneviva and  Correspondence and Footnotes
Correspondence and Footnotes
 P.R.B. Evora
P.R.B. Evora
Departamento de Cirurgia e Anatomia, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil
 References
References
Correspondence and Footnotes
Correspondence and Footnotes
Correspondence and Footnotes
Abstract
We investigated whether hepatic artery endothelium may be the earliest site of injury consequent to liver ischemia and reperfusion. Twenty-four heartworm-free mongrel dogs of either sex exposed to liver ischemia/reperfusion in vivo were randomized into four experimental groups (N = 6): a) control, sham-operated dogs, b) dogs subjected to 60 min of ischemia, c) dogs subjected to 30 min of ischemia and 60 min of reperfusion, and d) animals subjected to 45 min of ischemia and 120 min of reperfusion. The nitric oxide endothelium-dependent relaxation of hepatic artery rings contracted with prostaglandin F2a and exposed to increasing concentrations of acetylcholine, calcium ionophore A23187, sodium fluoride, phospholipase-C, poly-L-arginine, isoproterenol, and sodium nitroprusside was evaluated in organ-chamber experiments. Lipid peroxidation was estimated by malondialdehyde activity in liver tissue samples and by blood lactic dehydrogenase (LDH), serum aspartate aminotransferase (AST) and serum alanine aminotransferase (ALT) activities. No changes were observed in hepatic artery relaxation for any agonist tested. The group subjected to 45 min of ischemia and 120 min of reperfusion presented marked increases of serum aminotransferases (ALT = 2989 ± 1056 U/L and AST = 1268 ± 371 U/L; P < 0.01), LDH = 2887 ± 1213 IU/L; P < 0.01) and malondialdehyde in liver samples (0.360 ± 0.020 nmol/mgPT; P < 0.05). Under the experimental conditions utilized, no abnormal changes in hepatic arterial vasoreactivity were observed: endothelium-dependent and independent hepatic artery vasodilation were not impaired in this canine model of ischemia/reperfusion injury. In contrast to other vital organs and in the ischemia/reperfusion injury environment, dysfunction of the main artery endothelium is not the first site of reperfusion injury.
Key words: Endothelium, Hepatic artery, Nitric oxide, Liver, Ischemia/reperfusion injury
Introduction
Prolonged hepatic ischemia followed by reperfusion results in severe injury that contributes to the morbidity and mortality of liver transplantation. The liver damage occurring after the onset of reperfusion is a consequence of the interaction between different multifaceted mechanisms. It is widely accepted that nitric oxide (NO) is an essential mediator of liver injury. Recent observations have provided evidence that NO has beneficial effects on hepatic ischemia reperfusion (I/R) injury (1-4). Also, there is evidence that the earliest important event after reperfusion of an ischemic bed is a significant degree of endothelial dysfunction, characterized by the loss of NO release. Injury to a central artery seems to be the initial site of reperfusion injury in myocardial (5,6) and splanchnic I/R damage (7). The reduction in NO release may induce subsequent pathophysiologic phenomena observed in underlying tissue (low flow, vasospasm, thrombosis). In liver reperfusion injury, the failure of the microcirculation plays an important role, aggravating hepatic injury (8). The performance of the hepatic artery in the liver I/R injury has not been sufficiently studied. Thus, the specific aim of the present study was to determine whether the hepatic artery could be the initial site of reperfusion damage in a canine model of liver I/R injury.
Material and Methods
The study was carried out in accordance with the Guidelines of the University of São Paulo for Animal Experimentation and of the Animal Research Ethics Committee.
Animals
Twenty-four heartworm-free mongrel dogs of both sexes exposed to liver ischemia/reperfusion in vivo were randomized into four experimental groups (N = 6) a) control, sham-operated dogs; b) I60, dogs subjected to 60 min of ischemia; c) I30/R60, dogs subjected to 30 min of ischemia and 60 min of reperfusion, and d) I45/R120 animals subjected to 45 min of ischemia and 120 min of reperfusion.
Animal preparation
The 24 dogs of both sexes, fasted overnight and weighing 15-25 kg were sedated with intramuscular ketamine and anesthetized with intravenous pentobarbital sodium (25 mg/kg). Additional amounts were injected when necessary. Respiration was controlled with room air under mechanical ventilation. A rectal probe was used to monitor body temperature. A catheter was introduced into a superficial vein and saline (20 mL/h) was administered for fluid replacement. Blood pressure was monitored throughout the surgery with a catheter placed inside femoral artery. The abdomen was opened through a midline incision. The hepatic pedicle and hepatic artery (close to the celiac artery) were cross-clamped with vascular clamps. The collateral vessels of the hepatic artery were transected. The liver attachments were not divided, systemic heparinization was not applied and a side-to-side portocaval shunt was not constructed. Releasing the vascular clamps induced reperfusion. After the required I/R period, the animals were killed by exsanguination and the liver parenchyma was sampled, frozen and stored at -70ºC until the time of analysis.
In vitro experiments
The experiments were carried out immediately after exsanguination. The hepatic artery was dissected free of connective tissue and placed in Krebs solution of the following composition: 118.3 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 25.0 mM NaHCO3, 1.2 mM KH2PO4, 2.5 mM CaCl2, and 11.1 mM glucose. Hepatic artery rings (3-4 mm in length) were prepared with special care to avoid touching the intimal surface. The endothelium was removed by gently rubbing the intimal surface of the blood vessel with a watchmaker forceps in some of the rings in which vascular smooth muscle function was to be tested without the influence of the endothelium. This procedure removes the endothelium and conserves the ability of vascular smooth muscle to contract or relax. After their removal, hepatic artery rings were immediately placed in cold (7ºC) oxygenated Krebs solution. The rings were suspended in a classical eight-organ chambers setup (25 mL) filled with Krebs solution maintained at 37ºC and bubbled with 95% O2 and 5% CO2, pH 7.4. Two stainless steel clips passed through the vessel lumen suspended each ring. One clip was anchored to the bottom of the organ chamber, and the other was connected to a strain gauge for the measurement of isometric force (FT03, Grass Instrument Division, Astromed, Inc., West Warwick, RI, USA). The rings were placed at the optimal point of their length-tension relation (10 g) by being progressively stretched until maximal contraction in response to potassium ions (20 mM) at each level of distension. In all experiments, the presence or absence of endothelium was confirmed by determining the response to 1 µM acetylcholine (Ach) in rings contracted with 20 mM potassium ions. After optimal tension was achieved, the arterial rings were incubated with 1 µM and allowed to equilibrate for 60 min before drug administration. After equilibration was complete the rings were exposed to 2 µM prostaglandin F2a (PGF2a). Once a stable contraction was obtained, drugs were added to the bath at cumulative concentrations. After the cumulative response stabilized, the rings were washed and again allowed to equilibrate to baseline. The procedure was repeated with other drugs.
The following drugs were used: acetylcholine chloride, calcium ionophore (A23187), indomethacin, isoproterenol hydrochloride, phospholipase C (from C. perfringens), poly-L-arginine (molecular weight 139,200), PGF2a, sodium fluoride, and sodium nitroprusside (all from Sigma, St. Louis, MO, USA). All drugs were prepared daily with distilled water except for indomethacin, which was dissolved in Na2CO3 (10 µM), and calcium ionophore A32187, which was dissolved in dimethylsulfoxide (Sigma) and diluted further in distilled water. Concentrations were expressed as mM concentrations in the organ chamber. The NO release pathway and the pharmacological agonists for each step are illustrated in Figure 1.
Schematic diagram of the nitric oxide (NO) release pathway and the pharmacologic agonists used in the in vitro organ chamber studies. Ca A23187 = calcium ionophor; Ach = acetylcholine; NaF = sodium fluoride; PLC = phospholipase C; R = receptor; Gp = G-protein; PIP2 = inositol diphosphate; IP3 = inositol(1,4,5)-triphosphate; ISO = isoproterenol; SNP = sodium nitroprusside.
Aminotransferase assay
Blood was immediately centrifuged and serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH) were measured with a commercial kit (Labormed Co., Ltd., Guarulhos, SP, Brazil) and are reported as international units per liter.
Lipid peroxide assay
The levels of lipid peroxide in the liver were determined by a colorimetric reaction with thiobarbituric acid according to the method of Buege and Aust (9).
Statistical analysis
Data are reported as means ± SEM. In all experiments, N refers to the number of animals from which blood vessels were taken. In rings contracted with PGF2a, responses were expressed as percent change from the contraction levels. The in vitro vascular reactivity data were analyzed by analysis of variance (ANOVA) for paired or unpaired observations. The differences between specific mean malondialdehyde (MDA) values and serum enzymes determined by ANOVA were tested by post hoc analysis using the Student t-test with the Bonferroni correction. The Graphpad Prism 3.0 software for personal computers was applied for the statistical calculations. The level of significance was set at P < 0.05 in all analyses.
Results
Endothelium-dependent relaxation
The dose-response curves for Ach (a receptor agonist; 1 nM to 100 µM), sodium fluoride (a G-protein activator; 0.5 to 11.5 mM) are presented in Figure 2.
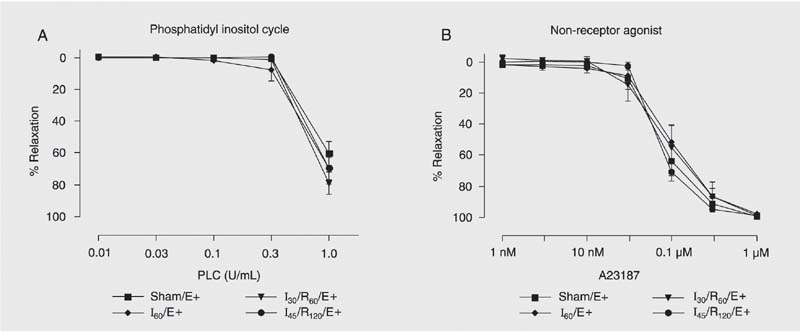
The dose-response curves for the cytosolic agonists phospholipase C (which induces phosphatidyl inositol hydrolysis; 0.01 to 1.0 U/mL), calcium ionophore A23187 (a non-receptor agonist; 1 nM to 1 µM) are presented in Figure 3.
The dose-response curves for poly-L-arginine (a hyperpolarization agent; 10 pM to 1 µM) are presented in Figure 4.
Similar endothelium-dependent relaxation was observed in hepatic artery rings from animals submitted to sham operation, ischemia, or I/R injury. There were no differences in the relaxation induced by these drugs among vascular rings from hepatic artery exposed to sham operation, 60 min of ischemia, 30 min of ischemia followed by 60 min of reperfusion, or 45 min of ischemia followed by 120 min of reperfusion.
Concentration-response curves to acetylcholine (Ach, A) and sodium fluoride (NaF, B) by the hepatic artery endothelial membrane. Hepatic artery segments were exposed to sham operation (Sham), 60 min of ischemia (I60), 30 min of ischemia followed by 60 min of reperfusion (I30/R60), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120) with endothelium (E+). Data are reported as means ± SEM, N = 6 per group. Arterial segments were first contracted with prostaglandin F2a and exposed to increasing drug concentrations. No significant difference was found among groups (ANOVA).
Concentration-response curves to phospholipase C (PLC, A) and calcium ionophore A23187 (B) by hepatic artery endothelial cells. Hepatic artery segments were exposed to sham operation (Sham), 60 min of ischemia (I60), 30 min of ischemia followed by 60 min of reperfusion (I30/R60), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120) with endothelium (E+). Data are reported as means ± SEM, N = 6 per group. Arterial segments were first contracted with prostaglandin F2a and exposed to increasing drug concentrations. No significant difference was found among groups (ANOVA).
Concentration-response curves to poly-L-arginine by hepatic artery segments. Hepatic artery segments were exposed to sham operation (Sham), 60 min of ischemia (I60), 30 min of ischemia followed by 60 min of reperfusion (I30/R60), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120) with endothelium (E+). Data are reported as means ± SEM, N = 6 per group. Arterial segments were first contracted with prostaglandin F2a and exposed to increasing drug concentrations. No significant difference was found among groups (ANOVA).
Endothelium-independent relaxation
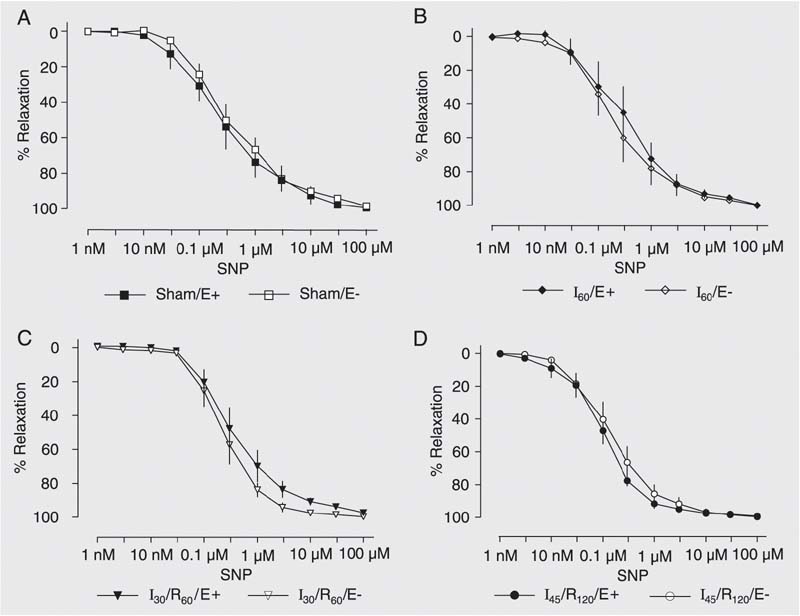
Isoproterenol (a ß-adrenergic agonist; 1 nM to 1 µM), and sodium nitroprusside (an NO donor; 1 nM to 100 µM) induced similar concentration-dependent relaxation of hepatic artery rings with or without endothelium from animals submitted to sham operation, 60 min of ischemia, 30 min of ischemia followed by 60 min of reperfusion, or 45 min of ischemia followed by 120 min of reperfusion (Figures 5 and 6).
Concentration-response curves to isoproterenol (ISO, 1 nM to 1 µM) by hepatic artery smooth muscle. Hepatic artery segments were exposed to sham operation (Sham, A), 60 min of ischemia (I60, B), 30 min of ischemia followed by 60 min of reperfusion (I30/R60, C), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120, D) with (E+) and without endothelium (E-). Data are reported as means ± SEM, N = 6 per group. Arterial segments were first contracted with prostaglandin F2a and exposed to increasing drug concentrations. No significant difference was found among groups (ANOVA).
Concentration-response curves to sodium nitroprusside (SNP, 1 nM to 100 µM) by hepatic artery segments exposed to sham operation (Sham, A), 60 min of ischemia (I60, B), 30 min of ischemia followed by 60 min of reperfusion (I30/R60, C), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120, D) with (E+) and without endothelium (E-). Data are reported as means ± SEM, N = 6 per group. Arterial segments were first contracted with prostaglandin F2a and exposed to increasing drug concentrations. No significant difference was found among groups (ANOVA).
Leakage of liver cell enzymes
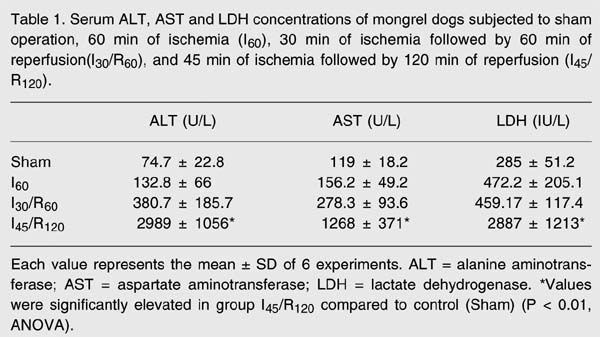
Hepatic injury was estimated by determining the release of ALT, AST and LDH (Table 1). Enzyme levels were significantly elevated in the group subjected to 45 min of ischemia followed by 120 min of reperfusion (I45/R120) compared to the groups subjected to sham operation, ischemia alone and 30 min of ischemia followed by 60 min of reperfusion (P < 0.01).
Serum ALT, AST and LDH concentrations of mongrel dogs subjected to sham operation, 60 min of ischemia (I60), 30 min of ischemia followed by 60 min of reperfusion (I30/R60), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120).
Liver lipid peroxide concentrations
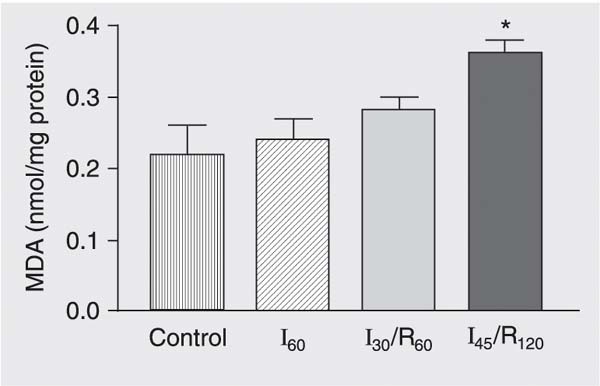
Liver lipid peroxide levels estimated by changes in MDA were significantly elevated in group I45/R120 compared with groups subjected to sham operation, ischemia alone and 30 min of ischemia followed by 60 min of reperfusion (P < 0.05; Figure 7).
Changes in malondialdehyde (MDA) concentrations in the liver of mongrel dogs exposed to sham operation (control), 60 min of ischemia (I60), 30 min of ischemia followed by 60 min of reperfusion (I30/R60), and 45 min of ischemia followed by 120 min of reperfusion (I45/R120). Data are reported as means ± SEM, N = 6 per group. Hepatic tissue lipid peroxide levels were significantly elevated in group I45/R120 compared to control.Each value represents the mean ± SEM of six experiments. *P < 0.05 compared to control (ANOVA).
Discussion
The purpose of the present study was to explore the initial time-course phase of liver and hepatic artery reperfusion injury. We chose a model of global ischemia (main hepatic artery cross-clamping) unmodified by any possible "protective interventions", so that we might isolate the effects of ischemia/reperfusion. This model is of near-global hepatic ischemia since the hepatic ligaments were not transected. The hepatic ischemia was induced without a portosystemic shunt in order to mimic operative conditions during major liver surgery in humans. Most animal models for studying hepatic ischemia involve partial occlusion of portal blood flow to avoid mesenteric congestion, which is poorly tolerated, but do not reflect the clinical scenario. The experiments were carried out using dogs because we are familiar with these animals from previous experiments in which coronary (10,11) and femoral arteries (12) were studied and presented endothelial dysfunction caused by ischemia followed by reperfusion.
Under the adopted experimental conditions there was no prostanoid-dependent vasorelaxation of the hepatic artery in any of the groups studied since all experiments were carried out in the presence of indomethacin.
Also there was no NO-dependent endothelial dysfunction observed in vitro, since ACh, sodium fluoride, phospholipase C, and calcium ionophore A23187 all induced statistically similar endothelium-dependent relaxation in the groups studied. Sodium nitroprusside and isoproterenol induced endothelium-independent vasorelaxation. Analysis from poly-L-arginine (a hyperpolarization agent) curves suggests that there was no non-nitric vasorelaxation impairment (13). Also, we tested poly-L-arginine and two endothelium-independent vasorelaxants: sodium nitroprusside (an NO donor with a cGMP-dependent direct vascular smooth muscle action) and isoproterenol (a ß-adrenergic agonist with a cAMP-dependent direct vascular smooth muscle action).
In the present study, periods of ischemia of at most 45 min were used before reperfusion was allowed in vivo. It is possible that under these conditions a longer time of ischemia may be required before reperfusion can induce the very early endothelial damage observed by others (5,6). The longest I/R time (I45/R120) was adopted on the basis of the enzymatic evidence of hepatic insufficiency and lipid peroxidation. A marked leakage of AST, ALT and LDH was observed in group I45/R120, reflecting hepatocyte damage and a significant MDA increase was observed in animals subjected to 45 min of ischemia and 120 min of reperfusion in vivo. MDA, a relatively crude product of cell membrane lipid peroxidation by superoxide radicals, was measured in liver tissue. A significant increase of lipid peroxide was observed in group I45/R120. Lipid peroxidation and MDA content in tissues have been related to irreversible ischemia and oxidative stress (14,15), although non-parenchymal cells rather than hepatocytes have been indicated to be the target for lipid peroxidation (14,16). The change in MDA values recorded in group I45/R120 indicates that there was enough time to permit oxidative stress and tissue damage in that group.
The reason why there were no differences in hepatic artery relaxation between groups is a matter of speculation. Discrepancies in endothelium function among different animal species as well as in different vessels from same species have been well documented (17).
Even assuming the effectiveness of "organ chamber" studies in detecting changes in endothelial cell responsiveness, the negative results do not allow us to conclude that hepatic ischemia/reperfusion injury occurs independently of endothelial dysfunction of the hepatic artery. First, the experiments were performed ex vivo and therefore it would still be possible that the responsiveness would be altered in vivo and might be transient in natura. Even if additional time points had been studied soon after the onset of reperfusion, there would still remain the possibility that the episodes of altered endothelial function, which might contribute to hepatic ischemia reperfusion damage, were missed.
The importance of the hepatic microcirculation in the pathophysiology of hepatic reperfusion injury is universally recognized (8). Failure of the hepatic microcirculation during reperfusion prolongs the period of hypoxia after the onset of reperfusion.
The contribution of the hepatic artery to hepatic reperfusion injury has not been sufficiently investigated. The vascular responses of livers perfused in vitro for up to 5 h have suggested that there is little deterioration in endothelial and smooth muscle function in the hepatic artery under these conditions (18). The hypothesis that the endothelium of central arteries of vital organs may be the earliest site of I/R injury, and that its dysfunction can induce pathophysiological phenomena also seems not to have been confirmed for the renal vasculature. Renal artery ring preparations from rabbit kidneys submitted to renal ischemia followed by reperfusion have demonstrated that the renal vasculature is relatively resistant to ischemia and probably is not involved in the physiopathology of postischemic renal failure (19).
The present study was designed to clarify the role of the hepatic artery in in vivo I/R injury. The liver has a double-blood supply: the hepatic artery coming from the celiac axis supplies the liver with arterial blood and the portal vein brings venous blood from the splanchnic vascular bed. From the portal vein the liver receives hepatotropic factors, including hormones, vasoactive substances, and other factors that can modify the hepatic I/R damage by some unknown mechanism. In the present experimental model, the liver was reperfused with splanchnic blood. These special characteristics make the liver a singular organ in the body. This fact can be an explanation for the apparent differences observed between the present study and observations made in coronary and splanchnic vascular beds, where endothelial dysfunction precedes I/R injury to the organs.
In conclusion, no abnormal changes in hepatic arterial vasoreactivity could be observed: endothelium-dependent and independent hepatic artery vasodilatation was not impaired in the canine model of I/R adopted here. The liver dysfunction caused by I/R was observed regardless of the presence of hepatic artery endothelium in vitro and the functional organ insult could be detected earlier than impaired hepatic artery endothelial function. Thus, normal function of the hepatic artery does not prevent liver insult in the ischemia-reperfusion environment.
Research supported by FAPESP and Fundação de Apoio ao Ensino, Pesquisa e Assistência (FAEPA) do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo (USP). Received November 25, 2006. Accepted April 9, 2007.
- 1. Koeppel TA, Thies JC, Schemmer P, Trauner M, Gebhard MM, Otto G, et al. Inhibition of nitric oxide synthesis in ischemia/reperfusion of the rat liver is followed by impairment of hepatic microvascular blood flow. J Hepatol 1997; 27: 163-169.
- 2. Cottart CH, Do L, Blanc MC, Vaubourdolle M, Descamps G, Durand D, et al. Hepatoprotective effect of endogenous nitric oxide during ischemia-reperfusion in the rat. Hepatology 1999; 29: 809-813.
- 3. Kobayashi H, Nonami T, Kurokawa T, Takeuchi Y, Harada A, Nakao A, et al. Role of endogenous nitric oxide in ischemia-reperfusion injury in rat liver. J Surg Res 1995; 59: 772-779.
- 4. Wang Y, Mathews WR, Guido DM, Farhood A, Jaeschke H. Inhibition of nitric oxide synthesis aggravates reperfusion injury after hepatic ischemia and endotoxemia. Shock 1995; 4: 282-288.
- 5. Mcfee MK, Hashimoto K, Pearson PJ, Schaff HV. Endothelial cell injury during global cardiac ischemia and reperfusion. Surg Forum 1990; 41: 301-311.
- 6. Tsao PS, Lefer AM. Time course and mechanism of endothelial dysfunction in isolated ischemic- and hypoxic-perfused rat hearts. Am J Physiol 1990; 259: H1660-H1666.
- 7. Carey C, Siegfried MR, Ma XL, Weyrich AS, Lefer AM. Antishock and endothelial protective actions of a NO donor in mesenteric ischemia and reperfusion. Circ Shock 1992; 38: 209-216.
- 8. Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. Am J Surg 2001; 181: 160-166.
- 9. Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol 1978; 52: 302-310.
- 10. Evora PR, Pearson PJ, Schaff HV. Impaired endothelium-dependent relaxation after coronary reperfusion injury: evidence for G-protein dysfunction. Ann Thorac Surg 1994; 57: 1550-1556.
- 11. Volpe MA, Carneiro JJ, Magna LA, Viaro F, Origuela EA, Evora PR. The role of magnesium in the endothelial dysfunction caused by global ischemia followed by reperfusion: in vitro study of canine coronary arteries. Scand Cardiovasc J 2003; 37: 288-296.
- 12. Joviliano EE, Piccinato CE, Cherri J, Viaro F, Evora PR. Inferior canine hindlimb ischemia and reperfusion impairs femoral artery endothelium-dependent wall relaxation. Vasc Endovascular Surg 2005; 39: 39-46.
- 13. Rodrigues AJ, Evora PR, Pearson PJ, Schaff HV. Endothelium-dependent relaxation to poly-L-arginine in canine coronary arteries. Endothelium 2003; 10: 79-87.
- 14. Ma NS. Changes in malondialdehyde contents in serum and tissues after ischemia and reperfusion of the bowel in dogs. Zhonghua Zheng Xing Shao Shang Wai Ke Za Zhi 1992; 8: 127-129.
- 15. Walsh TR, Rao PN, Makowka L, Starzl TE. Lipid peroxidation is a nonparenchymal cell event with reperfusion after prolonged liver ischemia. J Surg Res 1990; 49: 18-22.
- 16. Mathews WR, Guido DM, Fisher MA, Jaeschke H. Lipid peroxidation as molecular mechanism of liver cell injury during reperfusion after ischemia. Free Radic Biol Med 1994; 16: 763-770.
- 17. Piepot HA, Groeneveld AB, van Lambalgen AA, Sipkema P. Endotoxin impairs endothelium-dependent vasodilation more in the coronary and renal arteries than in other arteries of the rat. J Surg Res 2003; 110: 413-418.
- 18. Yang W, Benjamin IS, Sherwood R, Alexander B. Correlation of endothelium-dependent and -independent vasodilatation with liver function tests during prolonged perfusion of the rat liver. J Pharmacol Toxicol Methods 1998; 40: 227-234.
- 19. Pruneau D, Belichard P. Endothelium-dependent control of vascular tone in the rabbit kidney after ischemia and reperfusion. Eur J Pharmacol 1993; 231: 215-221.
Correspondence and Footnotes

Publication Dates
-
Publication in this collection
18 June 2007 -
Date of issue
June 2007
History
-
Accepted
09 Apr 2007 -
Received
25 Nov 2006