Resumo
An evaluation of the pesticides extraction from onion using a modern sample preparation method (QuEChERS) and determination by liquid chromatography tandem mass spectrometry was carried out. All the calibration curves showed r>0.99. The recoveries ranged between 61.8 and 120.0% with relative standard deviation lower than 20% for all compounds. Due to the occurrence of matrix effect, the quantification was performed using matrix-matched calibration. The limits of quantification of the method were between 0.0005 and 0.05 mg kg-1. The method shows the advantages of not require the clean-up step and consume low volume of organic solvents, decreasing time, costs and residues.
pesticides; QuEChERS; LC-ESI-MS/MS
pesticides; QuEChERS; LC-ESI-MS/MS
ARTIGO
Otimização e validação de método empregando quechers modificado e lc-esi-ms/ms para determinação de agrotóxicos em cebola
Optimization and validation of a method using modified quechers and lc-esi-ms/ms for the determination of pesticides in onion
Sherol Acosta RodriguesI; Sergiane Souza CaldasI; Eliana Badiale FurlongI; Ednei Gilberto PrimelI, * * e-mail: eprimelfurg@gmail.com ; Renato ZanellaII
IEscola de Química e Alimentos, Universidade Federal do Rio Grande, 96201-900 Rio Grande - RS, Brasil
IIDepartamento de Química, Universidade Federal de Santa Maria, 97105-900 Santa Maria - RS, Brasil
ABSTRACT
An evaluation of the pesticides extraction from onion using a modern sample preparation method (QuEChERS) and determination by liquid chromatography tandem mass spectrometry was carried out. All the calibration curves showed r>0.99. The recoveries ranged between 61.8 and 120.0% with relative standard deviation lower than 20% for all compounds. Due to the occurrence of matrix effect, the quantification was performed using matrix-matched calibration. The limits of quantification of the method were between 0.0005 and 0.05 mg kg-1. The method shows the advantages of not require the clean-up step and consume low volume of organic solvents, decreasing time, costs and residues.
Keywords: pesticides; QuEChERS; LC-ESI-MS/MS.
INTRODUÇÃO
A cebola (Allium cepa L) está entre os alimentos mais consumidos no mundo. Devido às propriedades antioxidantes e anticancerígenas, é considerada um alimento funcional, por consequência da presença de compostos bioativos, como as antocianinas e a quercetina em sua composição.1-3
O maior produtor de cebola do mundo é a China, responsável por cerca de 30% da produção mundial. Nessa mesma escala, o Brasil está em 9º lugar, como o maior produtor da América do Sul.4
No Brasil, a cebola ocupa a terceira posição em importância econômica, com plantio que se estende desde a região Sul até a região Nordeste. No estado do Rio Grande do Sul, o município do Rio Grande destaca-se com relação à produção.5
A cultura da cebola, contudo, é passível de ser atacada por várias doenças de origem fúngica, bacteriana, viral e nematológica, as quais podem ocorrer no campo, na pós-colheita, no transporte, no armazenamento e na comercialização. Como consequência dessa vulnerabilidade, a produtividade da cebola decresce drasticamente, podendo atingir até 100% de perda na produção de bulbos comercializáveis.6,7
Embora o uso de agrotóxicos seja necessário para garantir bons resultados de produtividade, o aumento da aplicação dos mesmos na cultura de cebola8 fez com que, em 2008, fosse incluída no Programa de Análise de Resíduos de Agrotóxicos em Alimentos (PARA), iniciado em 2001, pela Agência Nacional de Vigilância Sanitária (ANVISA), com vistas a avaliar continuamente os níveis de resíduos de agrotóxicos presentes nos alimentos in natura que chegam à mesa do consumidor. Das 103 amostras de cebola analisadas, 2,91% (3 amostras) foram consideradas insatisfatórias, em decorrência, exclusivamente, do uso de acefato, agrotóxico não autorizado para a cultura em estudo.9
Entre as dificuldades que envolvem as análises de agrotóxicos em amostras de alimentos como a cebola, destaca-se a complexidade dos procedimentos necessários para extração e purificação dessas amostras. A otimização das etapas envolvidas no preparo da mesma é essencial para reduzir o tempo e as fontes de erro relacionadas ao procedimento.10
Dentre as técnicas mais utilizadas para a extração de agrotóxicos em alimentos, podem ser citadas a extração acelerada por solvente,11 extração líquido-líquido,12 extração por ultrassom,13 extração com fluido supercrítico14,15 e a extração por dispersão da matriz em fase sólida.16-23
Nos últimos anos, técnicas e métodos de preparo de amostra baseados na minimização do uso de solventes orgânicos para a extração de agrotóxicos foram desenvolvidos como, por exemplo, o método QuEChERS (Quick, Easy, Cheap, Effective, Ruged and Safe), o qual tem sido largamente empregado para determinação de agrotóxicos em matrizes complexas. Foi assim denominado pelas características que possui: rapidez, facilidade, economia, efetividade, robustez e segurança. É baseado em uma extração com acetonitrila, seguida de partição líquido-líquido (adição de MgSO4 e NaCl) e posterior etapa de purificação com extração em fase sólida dispersiva.24-26 É um método robusto,27 adotado nos EUA, como o oficial da Association of Official Analytical Chemists para a determinação de resíduos de pesticidas em alimentos.28 É também considerado método oficial pelo European Committee for Standardization.26,29,30
Ao longo de seu desenvolvimento, o método QuEChERS tem sofrido variadas modificações, na direção de ser empregado como um método multirresíduo para extração de agrotóxicos em alimentos, usando técnicas cromatográficas acopladas à espectrometria de massas.20,31,32 Nesse contexto, o objetivo do presente estudo foi propor modificações no método QuEChERS, utilizado, no caso, para a extração de resíduos de agrotóxicos em cebola, empregando determinação por cromatografia líquida com fonte de ionização por eletronebulização acoplada à espectrometria de massas sequencial (liquid chromatogtaphy tandem mass spectrometry with electrospray ionization, LC-ESI-MS/MS).
PARTE EXPERIMENTAL
Escolha dos agrotóxicos
Os agrotóxicos foram selecionados a partir de entrevistas com produtores de cebola dos municípios de São José do Norte e Rio Grande, no estado do Rio Grande do Sul. Foram feitas perguntas a respeito dos agrotóxicos mais empregados na cultura de cebola, época de aplicação e a existência de orientação para aplicação dos mesmos.
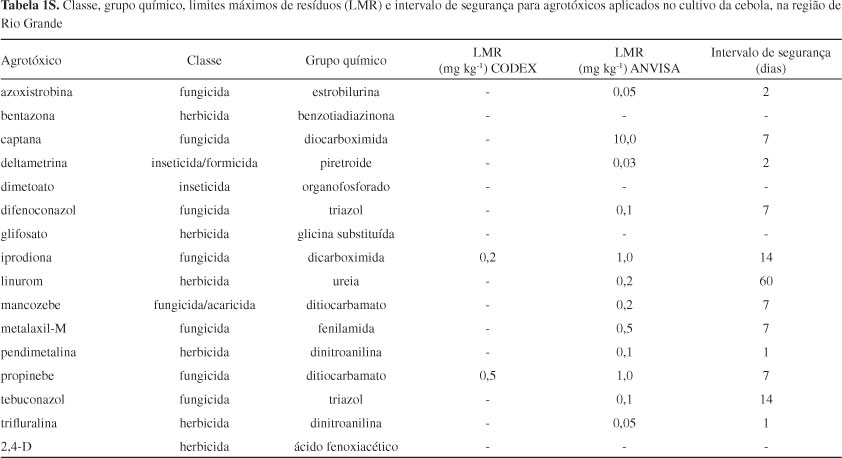
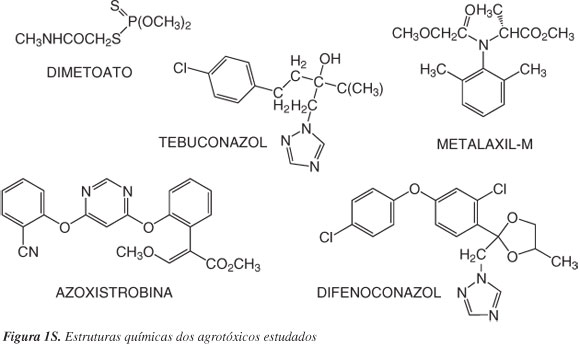
A partir dos dados obtidos, foi realizada uma pesquisa nas páginas eletrônicas da ANVISA e do Ministério da Agricultura, Pecuária e Abastecimento (MAPA)33,34 a fim de identificar quais das substâncias mais empregadas são permitidas para a cultura de cebolae quais poderiam ser determinadas no laboratório. Os agrotóxicos selecionados para o trabalho foram dimetoato, metalaxil-M, tebuconazol, azoxistrobina e difenoconazol. A Figura 1S, material suplementar, apresenta as estruturas químicas dos agrotóxicos estudados. A classe, o grupo químico, os limites máximos de resíduos (LMR) e o intervalo de segurança para agrotóxicos aplicados no cultivo da cebola, na região de Rio Grande, assim como as propriedades físico-químicas de cada substância são apresentados nas Tabelas 1S e 2S, material suplementar. O agrotóxico dimetoato, apesar de não ter LMR estabelecido para a cultura de cebola pela ANVISA, foi escolhido por ter o uso constantemente relatado nas entrevistas com os agricultores e, também, por sua elevada toxicidade.
Instrumentação
Foi utilizado um cromatógrafo a líquido Alliance Separations modelo 2695 Waters (Milford, MA, USA) equipado com amostrador automático, bomba quaternária, sistema de desgaseificação e coluna analítica Waters XTerra® MS C18 144 Å (50 × 3 mm, 3,5 µm - Milford, MA, USA), acoplado ao espectrômetro de massas Micromass® Quatro MicroTM API Waters, com fonte API, utilizando o modo de ionização por eletronebulização e sistema de aquisição de dados via software Masslynx 4.0 Waters.
Para o preparo das amostras de cebola, foi empregado um processador de alimentos, modelo Mega Master Plus RI 3170; Vórtex modelo Certomat® MV-B Braun (Bioteck Internacional, Alemmar - Comercial e Industrial S.A.) e centrífuga de tubos microprocessada modelo Quimis® Q222T (Quimis Aparelhos Científicos).
Reagentes
Os padrões analíticos dos agrotóxicos dimetoato (pureza > 99,4%), metalaxil-M (pureza > 99,0%), tebuconazol (pureza > 99,6%), azoxistrobina (pureza > 97%), difenoconazol (pureza > 99%) foram obtidos da Sigma-Aldrich (São Paulo, Brasil). O ácido fórmico grau analítico (pureza 98%) foi adquirido da Merck (Darmstadt, Alemanha).
Acetonitrila, grau cromatográfico, foi fornecida pela J.T. Baker (Mallinckrodt, Phillisburg, NJ, USA) e a água foi purificada em sistema Direct-Q UV3® (resistividade 18,2 MΩ cm, Millipore, USA). Sulfato de magnésio anidro e cloreto de sódio foram adquiridos da J.T. Baker; bondesil-PSA (40 µm) foi obtido da Varian (Palo Alto, CA, USA).
Preparo das soluções analíticas
As soluções analíticas estoque, contendo 1000 mg L-1 de cada agrotóxico foram preparadas pela dissolução do padrão sólido em metanol, considerando-se o grau de pureza. As soluções foram armazenadas em frasco âmbar e estocadas a -18 ºC.
A partir das soluções estoque de 1000 mg L-1 foram preparadas soluções estoque de concentrações de 100 mg L-1 de cada agrotóxico em metanol. A solução trabalho contendo a mistura dos analitos foi preparada na concentração de 10 mg L-1; foram realizadas diluições para a otimização dos parâmetros de fragmentação, preparo das curvas analíticas e nos ensaios da exatidão. A solução trabalho foi preparada mensalmente, enquanto as diluições utilizadas, diariamente.
Avaliação da vazão da fase móvel
A fase móvel empregada foi composta por água ultrapura e acetonitrila, acidificadas com 0,1% de ácido fórmico, na proporção (52:48, v/v). As vazões da fase móvel testadas foram de 0,2; 0,3 e 0,4 mL min-1. Os solventes empregados na fase móvel foram filtrados a vácuo, através de membranas de nylon 0,45 µm, e desgaseificados em banho de ultrassom durante 30 min, à temperatura ambiente.
Otimização dos parâmetros de fragmentação
A otimização dos parâmetros de fragmentação foi realizada com vistas a selecionar as melhores condições de fragmentação dos íons na análise por MS. Para tanto, foram realizadas infusões diretas no espectrômetro de massas com solução analítica na concentração de 1,0 mg L-1 de cada agrotóxico estudado. Os parâmetros variados foram: voltagem do cone, energia de colisão, temperatura da fonte, temperatura e vazão do gás de dessolvatação, voltagem do capilar e do cone extrator. A etapa de otimização das condições de fragmentação foi determinante para selecionar os íons a serem monitorados para qualificação e quantificação dos agrotóxicos estudados.
Otimização do procedimento de extração
Em 2008, um método empregando QuEChERS modificado para determinação de agrotóxicos em frutas e vegetais foi desenvolvido.32 Neste trabalho, foram realizadas modificações no método proposto, sendo realizados testes para avaliar a influência da etapa de purificação e da adição de cloreto de sódio na exatidão do método.
Avaliação da influência da etapa de purificação na exatidão do método
O procedimento foi realizado com a pesagem de 10,0 g de amostra processada em um tubo de polipropileno (capacidade 50,0 mL), seguida por etapa de fortificação no nível de 1,0 mg kg-1, realizada através da adição de volume conhecido de solução padrão (100,0 µL) à amostra sólida. Em seguida, 10,0 mL de acetonitrila foram adicionados, seguido por agitação manual (15 s) e agitação em vórtex (1 min). Após, foram adicionados 4,0 g de sulfato de magnésio anidro e repetida a etapa de agitação; finalmente, o tubo foi centrifugado a 5000 rpm durante 3 min. Para o procedimento sem a etapa de purificação, uma alíquota do extrato foi retirada e 10,0 µL injetados no sistema cromatográfico.
Para o procedimento com etapa de purificação, 6,0 mL do extrato foram transferidos para um tubo de polipropileno (capacidade 15,0 mL), contendo 150,0 mg de amina primária-secundária (primary secondary amine, PSA) e 950,0 mg de MgSO4, seguido pelas etapas de agitação e centrifugação. Uma alíquota do extrato final foi retirada e 10,0 µL foram injetados no sistema cromatográfico.
Avaliação da influência de NaCl na extração
Métodos empregados em trabalhos prévios utilizam sais como MgSO4 e NaCl. Assim, foram realizados testes comparando-se os resultados de recuperação, ao se empregar os dois sais ou apenas MgSO4.24,32 A adição de sais promove o efeito salting out, incrementando o nível de recuperação para analitos mais polares e diminui a solubilidade dessas substâncias na fase aquosa, bem como a quantidade de água na fase orgânica.26
Para a realização dos testes foram pesados 10,0 g de amostra em 6 tubos de polipropileno (capacidade 50,0 mL). Por sua vez, as amostras foram fortificadas no nível de 1,0 mg kg-1 e foram adicionados 10,0 mL de acetonitrila em cada tubo; em 3 tubos foram adicionados 4,0 g de MgSO4 e 1,0 g de NaCl e, nas três amostras restantes, foram adicionados apenas 4,0 g MgSO4. O restante do procedimento foi realizado sem a etapa de purificação.
Avaliação do efeito de matriz (EM)
Para avaliar o EM, foram injetadas no sistema cromatográfico uma solução padrão com a mistura dos agrotóxicos, na concentração de 1,0 mg L-1, diluída no extrato branco da matriz (extraído pelo método proposto) e uma outra solução padrão com a mistura dos agrotóxicos diluída com metanol. O cálculo do EM (%) foi baseado na bibliografia.35
Validação do método
O método foi validado, avaliando-se os seguintes itens: curva analítica, linearidade, limite de detecção (limit of detection, LOD), limite de quantificação (limit of quantification, LOQ), exatidão (recuperação) e precisão (repetibilidade e precisão intermediária), parâmetros sugeridos pelo Instituto Nacional de Metrologia, Normalização e Qualidade Industrial (INMETRO) e pela ANVISA.36,37
Curva analítica e linearidade
A linearidade do instrumento e do método foi avaliada pelas curvas analíticas através de padronização externa no solvente e por superposição da matriz com soluções analíticas nas concentrações de 0,0005; 0,005; 0,05; 0,5 e 1,0 mg L-1.
Para padronização externa por superposição da matriz, foram preparadas soluções analíticas através de diluições da solução padrão de trabalho com o extrato da matriz. Os procedimentos foram realizados com a matriz isenta de agrotóxicos e a padronização externa no solvente foi estabelecida por diluições da solução padrão de trabalho com metanol.
Cada solução foi injetada três vezes e os dados de regressão linear foram obtidos com auxílio do software do equipamento.
Limites de detecção e quantificação
Os limites de detecção (LODm) e quantificação (LOQm) do método foram obtidos pela injeção de soluções analíticas de diferentes concentrações, preparadas através de diluições da solução padrão de trabalho no extrato da matriz. O LODm e o LOQm para cada agrotóxico foram estimados a partir da relação sinal/ruído, calculada pelo software do equipamento, considerando, no mínimo, 3 e 10 vezes a razão do sinal pela linha de base (ruído), respectivamente.
Exatidão
A exatidão do método foi avaliada em função dos ensaios de recuperação, de acordo com as determinações da ANVISA e do International Conference Harmonisation.38 Amostras de cebola (isentas de agrotóxicos) foram fortificadas nos níveis de 0,0005; 0,005; 0,05 e 1,0 mg kg-1, a partir da adição de um volume conhecido de solução padrão de trabalho com a mistura dos agrotóxicos a 10,0 g de amostra.
Precisão
A precisão instrumental foi avaliada a partir de injeções sucessivas de solução analítica padrão, com a mistura dos agrotóxicos (n=7), e estimada através do coeficiente de variação (CV) com relação à média das áreas de todas as injeções.
A precisão do método foi avaliada em função da repetibilidade e da precisão intermediária. Para o estudo da repetibilidade, foi realizada a extração da amostra fortificada com solução analítica padrão e com a mistura dos agrotóxicos em diferentes níveis de fortificação, em triplicata; cada nível foi injetado três vezes. A partir das 9 determinações, foi calculado o CV(%).38 A precisão intermediária foi avaliada, assim como a repetibilidade, porém em diferentes dias e com diferentes analistas.
RESULTADOS E DISCUSSÃO
Avaliação da vazão da fase móvel
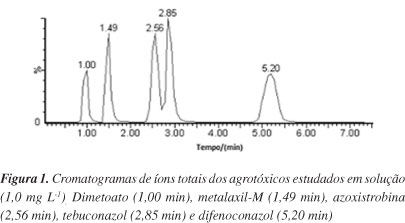
A melhor composição de fase móvel foi a mistura de acetonitrila e água ultrapura, acidificadas com 0,1% de ácido fórmico, na proporção de (52:48, v/v) e vazão de 0,4 mL min-1. Um cromatograma para a separação é apresentado na Figura 1. A acidificação permite que haja prótons suficientes na solução para que principalmente a espécie protonada esteja na solução, dissolvida na fase móvel, consequência (do controle) da distribuição de espécies. O pH menor proporciona maior interação entre as substâncias e a fase estacionária, mais apolar, aumenta a retenção da substância. Além disso, a acidificação da fase móvel também auxilia na ionização dos analitos.39,40
Ao se empregar a espectrometria de massas em série, através do modo de aquisição de monitoramento de reações múltiplas (MRM), é possível quantificar compostos que coeluem através do monitoramento das transições íon precursor - íon produto de cada substância. Assim, LC-MS/MS assegura a identificação com maior exatidão do que quando ela é feita apenas com base nas características de retenção das substâncias analisadas, como ocorre nas outras técnicas de detecção cromatográficas.41 Entretanto, a separação cromatográfica é importante para assegurar a eficiência da ionização dos analitos, diminuindo possíveis efeitos de matriz.35
Parâmetros de fragmentação otimizados
Foi utilizada a interface ESI, a mais indicada para substâncias neutras ou polares, que podem ser protononadas ou deprotonadas em condições apropriadas de pH.42
As melhores condições para a fragmentação dos íons monitorados, obtidas com infusões diretas no espectrômetro de massas na vazão de 10,0 µL min-1, foram: temperatura de 100 ºC na fonte; temperatura do gás N2 (gás de dessolvatação) de 350 ºC, mantida a uma vazão para a dessolvatação da amostra de 350 L h-1 e para o cone de seleção dos íons 50 L h-1; a energia do capilar foi 4 kV e no segundo cone, o extrator foi 3 V.
Na Tabela 1 são apresentadas as transições monitoradas no modo ESI (+), modo de aquisição MRM, energia de colisão, voltagem do cone e tempo de retenção dos agrotóxicos estudados.
Para cada analito foram selecionadas duas transições características, o que é possibilitado por um analisador de massas sequencial (tandem). A transição mais intensa (mais estável) foi escolhida para quantificação, enquanto a segunda, para confirmação.
Procedimento de extração
Avaliação da influência da etapa de purificação na exatidão do método
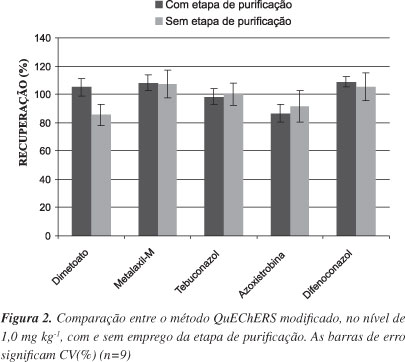
De acordo com os resultados de recuperação (R%) apresentados na Figura 2, é possível verificar que os valores de recuperação foram similares para a maioria dos agrotóxicos estudados com e sem a etapa de purificação. Para o primeiro caso, recuperações na faixa de 86,7 e 108,9% foram obtidas e, no segundo, as recuperações ficaram na faixa de 85,4 e 107,1%. Considerando que as possíveis interferências da matriz são compensadas pelo emprego de padronização externa por superposição da mesma, neste trabalho foi estabelecido o desenvolvimento do processo sem a etapa de purificação, a fim de tornar o procedimento de preparo da amostra ainda mais rápido e diminuir o consumo de reagentes.
Influência de NaCl na extração
Na Figura 3 são apresentados os resultados de recuperação, os quais indicam que a adição de cloreto de sódio em combinação ao sulfato de magnésio, conforme empregado em trabalhos prévios, gera valores de recuperação maiores para a maioria das substâncias quando comparados aos da adição de apenas MgSO4.24,32
Entretanto, para dimetoato, o valor de recuperação está acima de 150%, o que não é aceitável para a determinação de compostos traços em matrizes complexas e, além disso, MgSO4, quando empregado sozinho, é suficiente para gerar resultados de recuperação entre 70 e 120% para todos os analitos.
O valor elevado de recuperação para dimetoato pode ser explicado pelo fato de que o sal, além de favorecer a extração dos analitos, também aumenta a coextração de componentes da matriz, o que pode ter favorecido o enriquecimento do sinal. Assim, somente MgSO4 foi adicionado aos extratos, por possuir alta capacidade de remover água, quando comparado a outros sais.24
Efeito matriz
No método proposto foi constatado efeito matriz para o agrotóxico dimetoato. Essa influência se deve provavelmente à quantidade de interferentes presentes na amostra, ocasionando interferência na ionização dos analitos. Devido às interferências dos componentes da matriz no processo de ionização, a quantificação dos agrotóxicos foi realizada através de superposição da matriz. Na Figura 4, além do EM%, são apresentados resultados das recuperações dos agrotóxicos em diferentes formas de quantificação. Quando a recuperação é avaliada por comparação com as curvas no solvente, os valores variam de 49,0 a 105,0% com CV de 0,6 a 3,6% e, quando avaliada por meio da comparação por superposição na matriz, os valores para recuperação variaram de 89,6 a 114,1% com CV de 0,2 a 1,4%. O EM% variou de 28,0 a 100,0% com CV de 0,8 a 2,3%, comprovando, assim, a necessidade de quantificação dos agrotóxicos através de superposição da matriz.
Procedimento de extração otimizado
Na Tabela 2 são apresentadas as condições otimizadas para o método QuEChERS modificado, proposto no presente trabalho com relação a outros métodos, empregando QuEChERS, propostas em trabalhos anteriores.24,32 De acordo com os dados apresentados, é possível observar a diferença entre os métodos e afirmar que o proposto neste trabalho utiliza menor quantidade de reagentes e apresenta maior rapidez no procedimento de extração.
Validação do método
Curva analítica e linearidade
Através dos dados obtidos para a construção das curvas analíticas e análise das equações das retas no LC-ESI-MS/MS, é possível concluir que o modelo de regressão linear é adequado para as determinações analíticas em estudo. De acordo com a Tabela 3, os coeficientes de correlação (r) foram maiores que 0,99, estando de acordo com as orientações da ANVISA e do INMETRO, que recomendam, respectivamente, r > 0,99 e r > 0,90. 36,37
Limites de detecção e de quantificação
Os LODm e LOQm dos métodos foram de 0,016 e 0,05 mg kg-1 para o dimetoato; de 0,0016 e 0,005 mg kg-1 para o metalaxil-M e de 0,00016 e 0,0005 mg kg-1 para difenoconazol, azoxistrobina e tebuconazol.
Os limites de quantificação do método alcançaram valores menores que os limites máximos de resíduos estabelecidos pela ANVISA, para metalaxil-M, tebuconazol, azoxistrobina e difenoconazol. No que se refere ao agrotóxico dimetoato, não há LMR estabelecido para a cultura de cebola, por não ser indicado para aplicação na mesma.
Exatidão
Os processos mais utilizados para avaliar a exatidão de um método analítico são: materiais de referência, comparação de métodos, ensaios de recuperação e adição de padrão. Neste trabalho a exatidão foi avaliada por ensaios de recuperação.38 Para os ensaios de recuperação, diferentes níveis de concentração do analito foram empregados, visto que a eficiência do método pode variar em função da quantidade da substância adicionada.
A literatura sobre validação de métodos cromatográficos indica que os intervalos de recuperação aceitáveis para a determinação de resíduos devem estar entre 70 e 120%, com precisão de até ± 20%.38,40,43 Os valores de recuperação e de CV(%) são apresentados na Tabela 4. Os valores de recuperação variaram de 65,2 a 120,0%, com CV variando entre 3,3 e 20,0% para a repetibilidade e de 61,8 a 116,7%, com CV variando entre 2,0 e 20,0% para precisão intermediária em todos os níveis de fortificação.
Precisão
A precisão instrumental foi avaliada a partir de injeções sucessivas de solução analítica padrão na concentração de 0,05 mg L-1 (n=7). Os valores de CV(%) das áreas de todas as injeções foram: 10,5% para dimetoato; 6,7% para matelaxil-M; 7,8% para tebuconazol; 6,4% para azoxistrobina e 5,3% para difenoconazol.
A precisão do método foi avaliada em função da repetibilidade e da precisão intermediária, estimadas de acordo com as recomendações da ANVISA.37 Os valores de CV(%) para os estudos de repetibilidade (CVr) e precisão intermediária (CVpi) ficaram na faixa de 3,3-20,0% e 2,0-20,0%, respectivamente. Os valores mais detalhados de CVr e CVpi são apresentados, juntamente com os resultados de recuperação, na Tabela 4.
Para o dimetoato, os valores de CVr e CVpi foram estimados a partir de dois níveis de concentração. Isso porque o referido agrotóxico apresentou menor sensibilidade no sistema LC-ESI-MS/MS, relacionado às outras substâncias. A precisão do método foi avaliada em quatro níveis de fortificação. Os valores de CV para a repetibilidade e precisão intermediária, além de menores que 20%, ficaram muito próximos, indicando a adequada precisão do método.38
Aplicação do método
O método empregando QuEChERS modificado e LC-ESI-MS/MS, após ser validado, foi aplicado para determinação de resíduos dos agrotóxicos dimetoato, metalaxil-M, tebuconazol, azoxistrobina e difenoconazol. As amostras de cebola foram coletadas em um supermercado e em uma propriedade rural na região e resíduos de agrotóxicos não foram encontrados.
CONCLUSÕES
Os resultados obtidos neste estudo permitem concluir que o método empregando QuEChERS modificado e LC-ESI-MS/MS para a determinação dos agrotóxicos dimetoato, metalaxil-M, tebuconazol, azoxistrobina e difenoconazol em cebola é eficiente, rápido, preciso e exato.
O método QuEChERS modificado foi robusto e altamente reprodutível, uma vez que elevadas recuperações foram alcançadas. Além disso, o procedimento de extração é extremamente rápido e não necessita emprego da etapa de purificação dos extratos de cebola.
Entretanto, os componentes da matriz afetaram a eficiência da ionização de alguns analitos por LC-ESI-MS/MS e a influência do efeito matriz para dimetoato, metalaxil-M e azoxitrobina definiu o modo de quantificação dos analitos por calibração externa, através da superposição da matriz.
As condições cromatográficas e os parâmetros de fragmentação otimizados para determinação por LC-ESI-MS/MS permitiram a identificação e a quantificação dos agrotóxicos em estudo, em um tempo de análise menor que 6 min.
As curvas analíticas apresentaram valores de r maiores que 0,99 para as faixas de concentração necessárias às aplicações. Os valores médios de recuperação obtidos em diferentes níveis de fortificação estiveram na faixa de 61,8 a 120,0%, com valores de CV(%) menores que 20%. Os limites de quantificação do método variaram na faixa de 0,0005 a 0,05 mg kg-1.
O limite de quantificação do método para todos os analitos foi menor que os limites máximos de resíduos definidos pela legislação brasileira, ANVISA, para tais agrotóxicos em cebola. O fato é de suma importância, uma vez que o Brasil, em 2008, assumiu a liderança mundial no consumo de agrotóxicos, segundo dados do SINDAG; em função disso, no mesmo ano, o PARA incluiu a cebola em seu programa de monitoramento.
MATERIAL SUPLEMENTAR
Está disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre. A Tabela 1S apresenta a classe, o grupo químico, os LMR e o intervalo de segurança para agrotóxicos aplicados no cultivo da cebola, na região de Rio Grande. A Tabela 2S apresenta constante de acidez (pKa), coeficiente de partição octanol/água (Kow), solubilidade em água e pressão de vapor de cada substância para agrotóxicos em estudo e a Figura 1S, as estruturas químicas dos agrotóxicos estudados.
Recebido em 18/5/10; aceito em 3/12/10; publicado na web em 25/2/11
MATERIAL SUPLEMENTAR
- 1. Roldán, E.; Sánchez-Moreno, C.; Ancos, B.; Cano, M. P.; Food Chem. 2008, 108, 907.
- 2. Dini, I.; Tenore, G. C; Dini, A.; Food Chem. 2008, 107, 613.
- 3. Praksh, D.; Singh, B. N; Upadhyay, G.; Food Chem. 2007, 102, 1389.
-
4http://sistemasdeproducao.cnptia.embrapa.br/FontesHTML/Cebola/ CultivoCebolaNordeste/socioeconomia.htm, acessada em Outubro 2009.
» link -
5http://www.ibge.gov.br/cidadesat/topwindow.htm?1, acessada em Outubro 2009.
» link -
6http://www.cnph.embrapa.br/sistprod/cebola/caracteristicas_nutricionais.htm, acessada em Outubro 2009.
» link - 7. Soares, D. J.; Pitelli, R. A.; Braz, L. T.; Gravena, R.; Toledo, R. E. B.; Planta Daninha 2003, 21, 387.
- 8. Boff, P.; Debarba, J. F.; Silva, E.; Werner, H.; Horticultura Brasileira 2005, 23, 875.
-
9http://www.anvisa.gov.br/divulga/noticias/2009/pdf/150409_para.pdf, acessada em Outubro 2009.
» link - 10. Prestes, O. D.; Dissertação de Mestrado, Universidade Federal de Santa Maria, Brasil, 2007.
- 11. Chuang, J. C.; Hart, K.; Chang, J. S.; Boman, L. E.; Emon, J. M. V.; Reed, A. W.; Anal. Chim. Acta 2001, 444, 87.
- 12. Tahboub, Y. R.; Zaater, M. F.; Barri, T. A.; Anal. Chim. Acta 2006, 558, 62.
- 13. Rezic', I.; Horvat, A. J. M.; Babic', S.; Kastelan-Macan, M.; Ultrason. Sonochem. 2005, 12, 477.
- 14. Lehotay, S. J.; J. Chromatogr., A 1997, 785, 289.
- 15. Rissato, S. R.; Galhiane, M. S.; Knoll, F. R. N.; Apon, B. M.; J. Chromatogr., A 2004, 148, 143.
- 16. García-López, M.; Canosa, P.; Rodríguez, I.; Anal. Bioanal. Chem. 2008, 391, 963.
- 17. Rodrigues, S. A.; Caldas, S. S.; Primel, E. G.; Anal. Chim. Acta 2010, 678, 82.
- 18. Blasco, C.; Picó, Y.; Mañes, J.; Font, G.; J. Chromatogr., A 2002, 947, 227.
- 19. Dórea, H. D.; Lopes, W. G.; Quim. Nova 2004, 27, 892.
- 20. Wang, S.; Xu, Y.; Pan, C.; Jiang, S.; Liu, F.; Anal. Bioanal. Chem. 2007, 387, 673.
- 21. Wu, R.; Dang, Y.; Niu, L.; Hua, H.; J. Food Compos. Anal. 2008, 21, 582.
- 22. Yang, Y.; Shao, B.; Zhang, J.; Wu, Y.; Ying, J.; J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2008, 870, 241.
- 23. Pinho, G. P.; Neves, A. A.; Queiroz, M. E. L. R.; Quim. Nova 2009, 32, 92.
- 24. Anastassiades, M.; Lehotay, S. J.; Stajnbaher, D.; Schenck, F. J.; J. AOAC Int. 2003, 86, 412.
- 25. Romero-Gonzáles, R.; Frenich, A. G.; Vidal, J. L. M.; Talanta 2008, 76, 211.
- 26. Prestes, O. D.; Friggi, C. A.; Adaime, M. B.; Zanella, R.; Quim. Nova 2009, 32, 1620.
- 27. Lehotay, S. J.; J. AOAC Int. 2007, 90, 485.
-
28AOAC; Official Method 2007.01: Pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulphate, AOAC International, 2007.
-
29http://www.codexalimentarius.net, acessada em Outubro 2009.
» link -
30http://europa.eu/index_pt.htm, acessada em Outubro 2009.
» link - 31. Lehotay, S. J.; Kok, A.; Hiemstra, M.; Bodegraven, P. V.; J. AOAC Int. 2005, 88, 595.
- 32. Lesueur, C.; Knittl, P.; Gartner, M.; Mentler, A.; Fuerhacker, M.; Food Control 2008, 785, 289.
-
33http://www.anvisa.gov.br, acessada em Outubro 2009.
» link -
34http://agrofit.agricultura.gov.br/agrofit_cons/principal_agrofit_con, acessada em Outubro 2009.
» link - 35. Kruve, A.; Künnapas, A.; Herodes, K.; Leito, I.; Anal. Chim. Acta 2008, 1187, 58.
-
36Instituto Nacional de Metrologia, Normalização e Qualidade Industrial (INMETRO); Orientações sobre Validação de Métodos de Ensaios Químicos, DOQ-CGCRE-008. Revisão: 01, 2003.
-
37Agência Nacional de Vigilância Sanitária (ANVISA); Guia para Validação de Métodos Analíticos e Bioanalíticos, RE nº 889, de 29 de maio de 2003.
- 38. Ribani, M.; Bottoli, C. B. G.; Collins, C. H.; Jardim, I. C. S. F.; Melo, L. F. C.; Quim. Nova 2004, 27, 771.
- 39. Kuster, M.; Alda, M. L.; Barceló, D.; Mass Spectrom. Rev 2006, 25, 900.
- 40. Caldas, S. S.; Demoliner, A.; Costa, F. P.; D'Oca, M. G. M.; Primel, E. G.; J. Braz. Chem. Soc. 2010, 21, 642.
- 41. Chiaradia, M. C.; Collins, C. H.; Jardim, I. C. S. F.; Quim. Nova 2008, 31, 623.
- 42. Ardrey, R. E.; Liquid Chromatography - Mass Spectrometry: An Introduction; John Wiley & Sons Ltd, 2003, p. 276.
- 43. Brito, N. M.; Junior, O. P. A.; Polese, L.; Santos, T. C. R.; Ribeiro, M. L.; R. Ecotoxicol. e Meio Ambiente 2003, 13, 129.
Datas de Publicação
-
Publicação nesta coleção
18 Jul 2011 -
Data do Fascículo
2011
Histórico
-
Recebido
18 Maio 2010 -
Aceito
03 Dez 2010