Resumo
A Síndrome de Blau é uma doença de caráter hereditário autossômico dominante a qual também pode ocorrer de forma esporádica via mutação “de novo”. Em geral, tem aparecimento precoce ainda na primeira infância e sua tríade clássica inclui artrite, dermatite e uveíte. Este trabalho visa relatar as manifestações clínicas e principalmente oftalmológicas de uma paciente diagnosticada com Síndrome de Blau com ênfase ao achado incomum de infiltrados corneanos subepiteliais, raramente descrito na literatura.
Descritores:
Uveíte/etiologia; Uveíte/genética; Córnea; Artrite/genética; Mutação; Dermatite/genética; Criança
Abstract
The Blau syndrome is an autosomal dominant hereditary disease which can also occur sporadically via “de novo” mutation. Overall it has early onset and its classic triad includes arthritis, dermatitis and uveitis. This paper describes clinical and mainly especially ophthalmologic manifestations of a patient diagnosed with Blau syndrome with emphasis on an uncommon finding of corneal subepithelial infiltrates, rarely described in the literature.
Keywords:
Uveitis/etiology; Uveitis/genetics; Cornea; Arthritis/genetics; Dermatitis/genetics; Child
Introdução
A Sindrome de Blau é uma afecção de caráter hereditário autossômico dominante que ocorre, em 50-90% dos casos via mutação “de novo”.(11 Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014 ;12(1):33.) A manifestação da síndrome pode ser precoce e apresentar uma tríade clássica, incluindo artrite, dermatite e uveíte.(22 Caso F, Galozzi P, Costa L, Sfriso P, Cantarini L, Punzi L. Autoinflammatory granulomatous diseases: from Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn's disease. RMD Open. 2015;1(1):e000097.)
Em 1985, Blau e colaboradores descreveram uma nova síndrome caracterizada por artrite, uveíte anterior e rash cutâneo de intensidade e início variáveis em 11 membros de uma mesma família distribuídos em 4 gerações.(33 Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107(5):689-93.) Achados semelhantes também foram descritos por Jabs e colaboradores, quase simultaneamente.(44 Jabs DA, Houk JL, Bias WB, Arnett FC. Familial granulomatous synovitis, uveitis, and cranial neuropathies. Am J Med. 1985;78(5):801-4.) Em 1989, Pastores e colaboradores descreveram 3 casos caracterizados como granulomatose sistêmica juvenil de caráter familiar.(55 Pastores GM, Michels VV, Stickler GB, Su WP, Nelson AM, Bovenmyer DA. Autosomal dominant granulomatous arthritis, uveitis, skin rash, and synovial cysts. J Pediatr. 1990;117(3):403-8.) Os sinais e sintomas começam na infância, geralmente antes dos 4 anos de idade, podendo haver dermatite granulomatosa, que é geralmente o primeiro sinal da síndrome de Blau, artrite e uveíte. Posteriormente, com a identificação da mutação causadora da doença no NOD2 (66 Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19-20.,77 Raiji VR, Miller MM, Jung LK. Uveitis in Blau syndrome from a de novo mutation of the NOD2/CARD15 gene. J AAPOS. 2011;15(2):205-7.) uma nova linha de pesquisa se iniciou e foi possível perceber que a patologia pode ter tanto caráter familiar quanto esporádico.(88 Kanazawa N, Matsushima S, Kambe N, Tachibana T, Nagai S, Miyachi Y. Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J Invest Dermatol. 2004;122(3):851-2.) O diagnóstico é feito mediante a conjugação da clínica com a história familiar, detecção histológica e investigação laboratorial. Este relato descreve o caso de infiltrados subepiteliais corneanos à biomicroscopia, um achado ocular raro nessa síndrome.
Relato de Caso
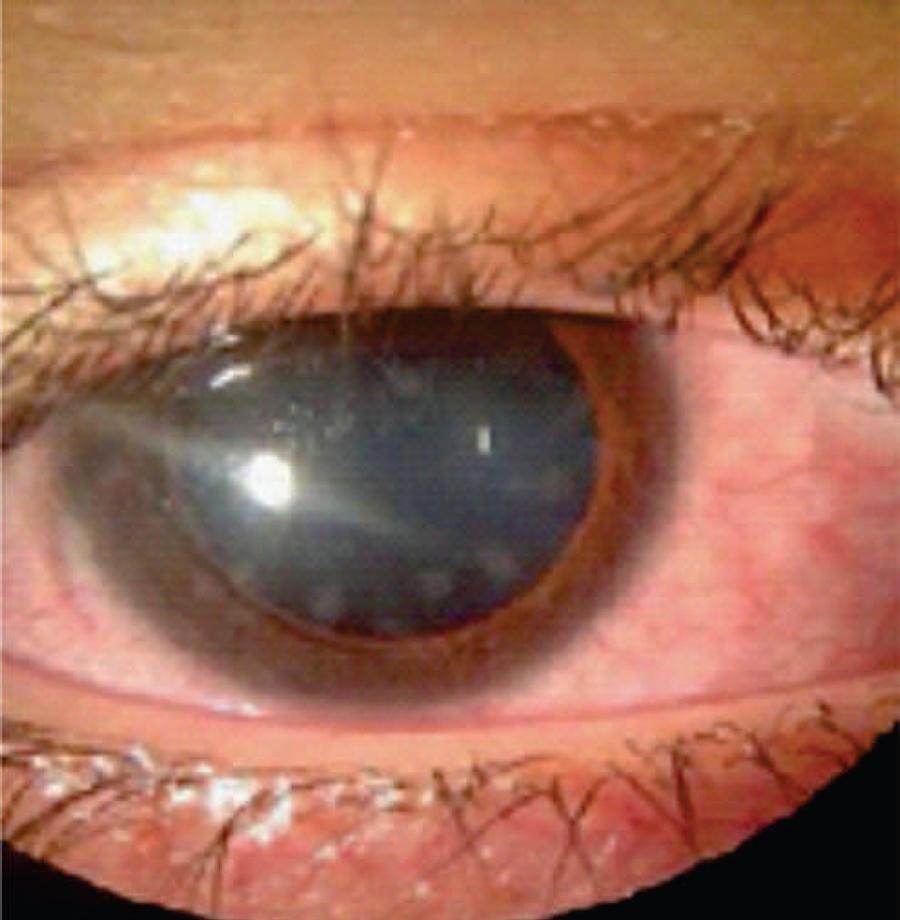
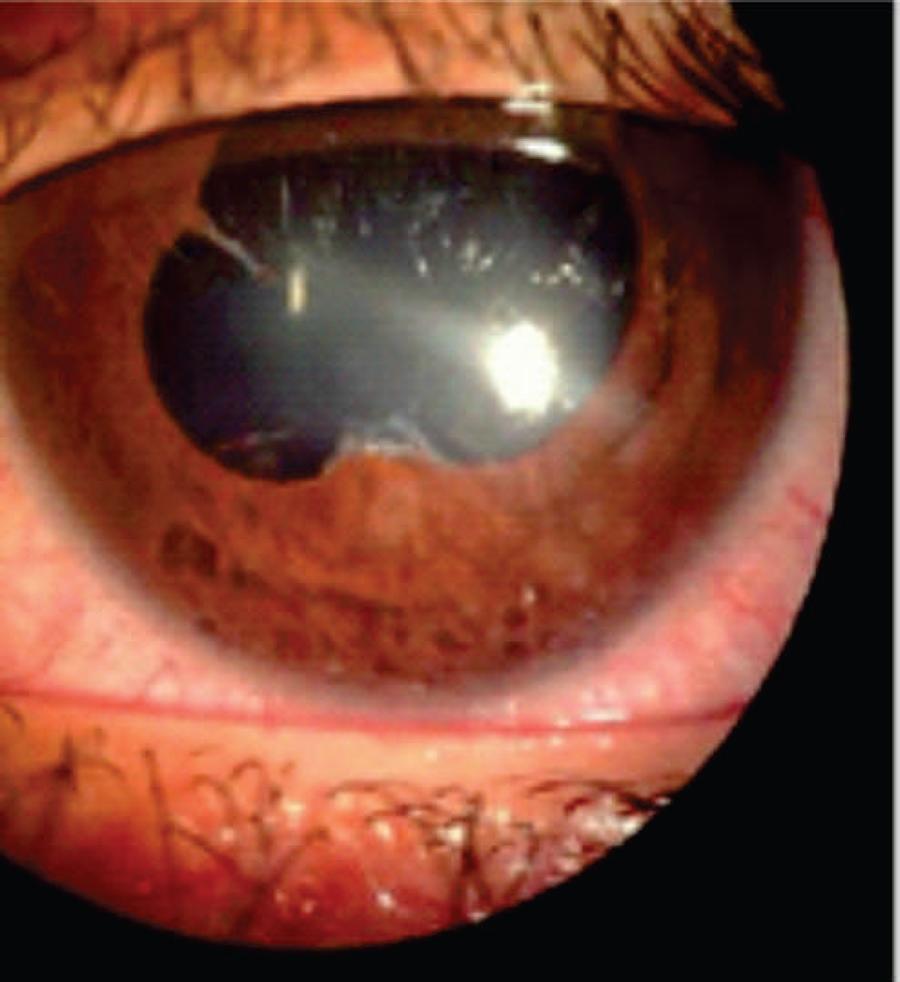
VVF, 13 anos, sexo feminino, apresentou aos 2 meses de idade exantema em face, membros e flancos, evoluindo progressivamente com artrite de mãos e punhos. Aos 3 anos, foi referenciada para exame oftalmológico queixando-se de hiperemia ocular e lacrimejamento. O exame revelou uveíte anterior em ambos os olhos (AO) sem outras alterações de risco. Os exames laboratoriais revelaram aumento da velocidade de hemossedimentação (VHS) e da proteína C reativa (PCR); fator reumatóide (FR) positivo e fator antinuclear (FAN) negativo. Para elucidação diagnóstica foi realizada biópsia cutânea que demonstrou a presença de múltiplos granulomas na derme sem sinais de necrose. O teste genético revelou presença da mutação R334W. Os pais também foram testados para a mutação com resultado negativo. A paciente foi tratada com prednisona, naproxeno e metotrexato e o tratamento oftalmológico incluiu o uso de colírios de acetato de prednisolona 1,0% e tropicamida 1,0% com bom controle das manifestações sistêmicas e oculares. Um ano após, apresentou recorrência dos sintomas sistêmicos e oculares e foi iniciado infliximabe com bons resultados. A paciente se encontra assintomática em uso de infliximabe, metotrexato e ácido fólico. O exame oftalmológico revelou acuidade visual igual a 20/20 em AO, a biomicroscopia revelou ausência de reação inflamatória, infiltrados subepiteliais ovóides em AO (Figuras 1 e 2) e sinéquias posteriores no olho esquerdo (Figura 2) e a fundoscopia revelou exame normal em AO.
Discussão
A Síndrome de Blau é uma doença de caráter hereditário autossômico dominante quando familiar, mas que pode, contudo, ocorrer de forma esporádica via mutação “de novo”.(77 Raiji VR, Miller MM, Jung LK. Uveitis in Blau syndrome from a de novo mutation of the NOD2/CARD15 gene. J AAPOS. 2011;15(2):205-7.) Sua prevalência permanece desconhecida, uma vez que muitos casos não são diagnosticados ou são enquadrados como outras doenças inflamatórias. Em geral os sintomas se iniciam antes dos quatro anos de vida e estão relacionados a uma mutação no gene NOD2.(99 Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54(10):3337-44.) A maioria das mutações relacionadas à Síndrome de Blau relatadas até a presente data estão no ou perto do domínio NOD / NACHT de ligação ao nucleotídeo da proteína NOD2, ainda que também possa ser encontrada na região C-terminal caracterizado por uma estrutura de repetição rica em leucina. Ao longo dos últimos anos houve um aumento do número de mutações diferentes NOD2 associados com Blau e novas mutações NOD2 ainda são catalogadas regularmente.(1010 Milman N, Ursin K, Rødevand E, Nielsen FC, Hansen TV. A novel mutation in the NOD2 gene associated with Blau syndrome: a Norwegian family with four affected members. Scand J Rheumatol. 2009;38(3):190-7.
11 Okada S, Konishi N, Tsumura M, Shirao K, Yasunaga S, Sakai H, et al. Cardiac infiltration in early-onset sarcoidosis associated with a novel heterozygous mutation, G481D, in CARD15. Rheumatology (Oxford). 2009;48(6):706-7.
12 Priori R, Bombardieri M, Spinelli FR, Merlin F, Miceli-Richard C, La Cava M, et al. Sporadic Blau syndrome with a double CARD15 mutation. Report of a case with lifelong follow-up. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21(3):228-31.
13 Sakai H, Ito S, Nishikomori R, Takaoka Y, Kawai T, Saito M, et al. A case of early-onset sarcoidosis with a six-base deletion in the NOD2 gene. Rheumatology (Oxford). 2010;49(1):194-6.
14 van Duist MM, Albrecht M, Podswiadek M, Giachino D, Lengauer T, Punzi L, et al. A new CARD15 mutation in Blau syndrome. Eur J Hum Genet. 2005;13(6):742-7.-1515 Villanueva-Mendoza C, Arellanes-García L, Cubas-Lorenzo V, Jimenez-Martinez MC, Flores-Suárez LF, Zenteno JC. Familial case of Blau syndrome associated with a CARD15/NOD2 mutation. Ophthalmic Genet. 2010;31(3):155-8.) No presente relato a paciente apresentou a mutação R334W, a qual é relacionada ao gene NOD2, sendo essa mutação responsável por 50-90% dos pacientes que apresentam a tríade clássica da Síndrome de Blau.
A doença é caracterizada por uma tríade clássica que inclui dermatite granulomatosa, artrite e uveíte. As lesões cutâneas geralmente aparecem dentro do primeiro ano de vida e o espectro pode variar desde rash simples a nódulos ou pápulas de caráter assintomático. Os primeiros sinais de artrite ocorrem entre 2 e 4 anos de vida e afeta tipicamente articulações periféricas, principalmente nos pulsos, joelhos, tornozelos e interfalangeana proximal das mãos. Tenossinovite é igualmente uma característica, assim como a presença de edema articular.(1616 Aróstegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805-13.) Outros sinais significativos no exame osteoarticular são cistos sinoviais e camptodactilia.(55 Pastores GM, Michels VV, Stickler GB, Su WP, Nelson AM, Bovenmyer DA. Autosomal dominant granulomatous arthritis, uveitis, skin rash, and synovial cysts. J Pediatr. 1990;117(3):403-8.)
Cerca de 80% dos pacientes desenvolveram doença ocular com início em média aos 4 anos de idade.(99 Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54(10):3337-44.) A uveíte pode se apresentar como uveíte anterior granulomatosa com potencial evolução para panuveíte grave com coroidite multifocal, sendo geralmente bilateral e frequentemente associada à perda visual severa. Nestes casos, a inflamação vítrea é comum e pode permanecer persistentemente ativa durante anos.(1717 Lindsley CB, Godfrey WA. Childhood sarcoidosis manifesting as juvenile rheumatoid arthritis. Pediatrics. 1985;76(5):765-8.) As manifestações oftalmológicas foram bem determinadas pelo estudo de Latkany e colaboradores,(1818 Latkany PA, Jabs DA, Smith JR, Rosenbaum JT, Tessler H, Schwab IR, et al. Multifocal choroiditis in patients with familial juvenile systemic granulomatosis. Am J Ophthalmol. 2002;134(6):897-904.) que revisou 16 pacientes de 8 famílias com granulomatose sistêmica juvenil. Destes, 14 pacientes apresentaram panuveíte com coroidite multifocal. Um paciente teve apenas uveíte anterior. Um paciente apresentou panuveíte e neuropatia óptica isquêmica provavelmente secundária à vasculite. Catarata, glaucoma, ceratopatia em faixa, edema macular cistide e edema do disco óptico foram descritos. Todos os pacientes apresentavam poliartrite e pelo menos 9 tiveram erupções cutâneas. No presente relato foi descrita uveíte anterior com envolvimento corneano bilateral, sem manifestações no segmento posterior ou complicações oftalmológicas secundárias.
Outras alterações sistêmicas presentes em pacientes portadores de Síndrome de Blau incluem febre, linfadenopatia, eritema nodoso, vasculite leucocitoclástica, neuropatias transitórias, nefrite intersticial glomerular granulomatosa com evolução potencial para insuficiência renal crônica, hipertensão arterial, pericardite e granulomas hepáticos.(1919 Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J, et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum. 2009;60(6):1797-803.) No presente relato, a paciente apresentou doença cutânea, oftalmológica e osteoarticular sem outras alterações sistêmicas.
Alterações laboratoriais comuns incluem hipercalcemia, hipercalciúria, aumento da enzima conversora de angiotensina (ECA) e da VHS, leucopenia, proteinúria, hematúria e piúria. As provas de função hepática podem estar alteradas e em geral o fator reumatoide FR é positivo.(99 Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54(10):3337-44.) Alterações laboratoriais foram encontradas no presente relato.
O exame histopatológico revela uma inflamação granulomatosa na derme que contêm células epitelióides e ocasionalmente células de Langerhans. A presença de infiltrado linfomononuclear é frequente e em nenhum caso foi observado a presença de necrose.(2020 Janssen CE, Rose CD, De Hertogh G, Martin TM, Bader Meunier B, Cimaz R, et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J Allergy Clin Immunol. 2012;129(4):1076-84.)
O diagnóstico é clínico a partir da identificação da tríade clássica e sintomas associados. A biópsia da pele deve ser realizada de forma complementar, considerando que a pele ofereceu melhor capacidade diagnóstica quando comparada a sinóvia, com histologia de confirmação em mais de 90% dos pacientes suspeitos de síndrome de Blau,(99 Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54(10):3337-44.) tal como no presente relato.
O tratamento sistêmico envolve o uso de corticosterides sistêmicos em doses variáveis, os quais devem ser utilizados com parcimônia uma vez que os efeitos colaterais do uso prolongado representam grande morbidade aos pacientes.(2121 Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509-16. Review.) Caso o controle da doença necessite de uso prolongado de corticosteroides ou o paciente apresente eventos adversos a este tratamento, é mandatório iniciar o uso de imunossupressores como o metotrexato e agentes biológicos anti-fator de necrose tumoral podem ser usados em conjunto ou separadamente, a fim de obter adequado controle da síndrome. A uveíte deve ser tratada de acordo com a classificação anatômica e o grau de inflamação.(2121 Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509-16. Review.)
Raramente pacientes com Síndrome de Blau apresentam infiltrados corneanos subepiteliais como o caso descrito de acordo com a literatura disponível até a presente data. Quando presentes, esses infiltrados são caracteristicamente ovides e não causam repercussão visual a não ser que obstruam o eixo visual.(77 Raiji VR, Miller MM, Jung LK. Uveitis in Blau syndrome from a de novo mutation of the NOD2/CARD15 gene. J AAPOS. 2011;15(2):205-7.)
Este relato contribui para a descrição de manifestações oftalmológicas incomuns da síndrome de Blau, uma vez que infiltrados corneanos ovóides são raramente observados.
-
Instituição: Departamento de Oftalmologia, Universidade Federal do Rio de Janeiro (UFRJ)
Referências
-
1Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014 ;12(1):33.
-
2Caso F, Galozzi P, Costa L, Sfriso P, Cantarini L, Punzi L. Autoinflammatory granulomatous diseases: from Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn's disease. RMD Open. 2015;1(1):e000097.
-
3Blau EB. Familial granulomatous arthritis, iritis, and rash. J Pediatr. 1985;107(5):689-93.
-
4Jabs DA, Houk JL, Bias WB, Arnett FC. Familial granulomatous synovitis, uveitis, and cranial neuropathies. Am J Med. 1985;78(5):801-4.
-
5Pastores GM, Michels VV, Stickler GB, Su WP, Nelson AM, Bovenmyer DA. Autosomal dominant granulomatous arthritis, uveitis, skin rash, and synovial cysts. J Pediatr. 1990;117(3):403-8.
-
6Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29(1):19-20.
-
7Raiji VR, Miller MM, Jung LK. Uveitis in Blau syndrome from a de novo mutation of the NOD2/CARD15 gene. J AAPOS. 2011;15(2):205-7.
-
8Kanazawa N, Matsushima S, Kambe N, Tachibana T, Nagai S, Miyachi Y. Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J Invest Dermatol. 2004;122(3):851-2.
-
9Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54(10):3337-44.
-
10Milman N, Ursin K, Rødevand E, Nielsen FC, Hansen TV. A novel mutation in the NOD2 gene associated with Blau syndrome: a Norwegian family with four affected members. Scand J Rheumatol. 2009;38(3):190-7.
-
11Okada S, Konishi N, Tsumura M, Shirao K, Yasunaga S, Sakai H, et al. Cardiac infiltration in early-onset sarcoidosis associated with a novel heterozygous mutation, G481D, in CARD15. Rheumatology (Oxford). 2009;48(6):706-7.
-
12Priori R, Bombardieri M, Spinelli FR, Merlin F, Miceli-Richard C, La Cava M, et al. Sporadic Blau syndrome with a double CARD15 mutation. Report of a case with lifelong follow-up. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21(3):228-31.
-
13Sakai H, Ito S, Nishikomori R, Takaoka Y, Kawai T, Saito M, et al. A case of early-onset sarcoidosis with a six-base deletion in the NOD2 gene. Rheumatology (Oxford). 2010;49(1):194-6.
-
14van Duist MM, Albrecht M, Podswiadek M, Giachino D, Lengauer T, Punzi L, et al. A new CARD15 mutation in Blau syndrome. Eur J Hum Genet. 2005;13(6):742-7.
-
15Villanueva-Mendoza C, Arellanes-García L, Cubas-Lorenzo V, Jimenez-Martinez MC, Flores-Suárez LF, Zenteno JC. Familial case of Blau syndrome associated with a CARD15/NOD2 mutation. Ophthalmic Genet. 2010;31(3):155-8.
-
16Aróstegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805-13.
-
17Lindsley CB, Godfrey WA. Childhood sarcoidosis manifesting as juvenile rheumatoid arthritis. Pediatrics. 1985;76(5):765-8.
-
18Latkany PA, Jabs DA, Smith JR, Rosenbaum JT, Tessler H, Schwab IR, et al. Multifocal choroiditis in patients with familial juvenile systemic granulomatosis. Am J Ophthalmol. 2002;134(6):897-904.
-
19Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J, et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum. 2009;60(6):1797-803.
-
20Janssen CE, Rose CD, De Hertogh G, Martin TM, Bader Meunier B, Cimaz R, et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J Allergy Clin Immunol. 2012;129(4):1076-84.
-
21Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509-16. Review.
Datas de Publicação
-
Publicação nesta coleção
Jan-Feb 2019
Histórico
-
Recebido
23 Mar 2018 -
Aceito
19 Nov 2018