Abstracts
Replacement schedules with hydrocortisone (HC) to treat 21OHD are generally unsatisfactory and partially successful regarding growth. Noncompliance is common since its short half-life requires TID administration. Even multiple daily HC doses do not reproduce cortisol chronobiology and may disturb hypothalamic-mediated rhythms. Because synthetic glucocorticoids could improve clinical control, we evaluated the possible benefits of a one-year treatment period with a single morning oral dose of prednisolone (PD) phosphate in 44 patients with 21OHD randomized to two sex and age-matched groups: one (n=23) receiving PD (2.4-3.5mg/m² BSA) and the other (n=21) TID HC (10-15mg/m² BSA). After one year, bone maturation ratio was kept stable in the PD group (from 1.20 to 1.14), whereas a slight increase was seen in the HC group (from 1.21 to 1.29). Growth velocity (SDS) was preserved in the PD group (from 1.2 to 1.2 in all; 0.79 to1.13 in pre-pubertals), whereas a slight increase occurred in the pre-pubertal HC-treated patients (from 1.1 to 1.9); height SDS for BA increased significantly in the PD group. Thus, patients with 21OHD treated for one year with a single morning dose of PD appear to achieve a better clinical and hormonal control than those on TID HC, permitting a reduction of the replacement dose. The current PD schedule used by our group (1.5-3mg/m² BSA/day) suggests a higher HC:PD bioequivalence ratio of 6-8:1.
21-Hydroxylase deficiency; Prednisolone; Hydrocortisone; Growth; Androgens
Esquemas de reposição com hidrocortisona (HC) para a D21OH são, em geral, insatisfatórios e parcialmente eficientes quanto ao crescimento. Não aderência é comum já que a meia-vida da HC requer administração de 3 doses diárias. Mesmo várias doses não reproduzem a cronobiologia do cortisol e podem prejudicar os ritmos mediados pelo hipotálamo. Como glicocorticóides sintéticos podem melhorar o controle clínico, avaliamos os possíveis benefícios da terapia por um ano com fosfato de prednisolona (PD) em dose única matinal em 44 pacientes com D21OHD randomizados em 2 grupos: um (n=23) recebeu PD (2,4-3,5mg/m²SC) e o outro (n=21), HC 3 vezes ao dia (10-15mg/m²SC). Após um ano, a taxa de maturação óssea manteve-se estável no grupo PD (de 1,20 para 1,14), tendo aumentado discretamente (de 1,21 para 1,29) no grupo HC. A velocidade de crescimento (em SDS) foi preservada no grupo PD (de 1,2 para 1,2 no total de pacientes; 0,79 para 1,13 nos pré-púberes), enquanto discreto aumento ocorreu nos pré-púberes sob HC (1,1 to 1,9); a estatura para a IO (em SDS) elevou-se após um ano do uso de PD. Assim, pacientes com D21OH tratados por um ano com dose única matinal de PD parecem alcançar melhor controle clínico e hormonal do que aqueles usando 3 doses diárias de HC, permitindo inclusive a redução da dose. Nosso esquema atual de reposição com PD (1,5-3mg/m²SC/dia), sugere uma relação de bioequivalência HC:PD de 6-8:1.
Deficiência de 21-hidroxilase; Prednisolona; Hidrocortisona; Crescimento; Andrógenos
ARTIGO ORIGINAL
One-year clinical evaluation of single morning dose prednisolone therapy for 21-hydroxylase deficiency
Avaliação clínica por um ano de terapia com prednisolona em dose única matinal para a deficiência de 21-hidroxilase
Milena C. F. Caldato; Vânia T. Fernandes; Claudio E. Kater
Adrenal and Hypertension Unit, Division of Endocrinology, Department of Medicine Federal University of São Paulo - UNIFESP/EPM, São Paulo,SP, Brazil
Correspondence Correspondence to Claudio E. Kater Steroids Laboratory, Adrenal and Hypertension Unit Division of Endocrinology, Department of Medicine Federal University of São Paulo - UNIFESP/EPM R. Pedro de Toledo, 781 13 th floor 04039-032 São Paulo, SP - Brazil Telefax: (5511) 5574-6502 E -mail: kater@endocrino.epm.br
ABSTRACT
Replacement schedules with hydrocortisone (HC) to treat 21OHD are generally unsatisfactory and partially successful regarding growth. Noncompliance is common since its short half-life requires TID administration. Even multiple daily HC doses do not reproduce cortisol chronobiology and may disturb hypothalamic-mediated rhythms. Because synthetic glucocorticoids could improve clinical control, we evaluated the possible benefits of a one-year treatment period with a single morning oral dose of prednisolone (PD) phosphate in 44 patients with 21OHD randomized to two sex and age-matched groups: one (n=23) receiving PD (2.4-3.5mg/m2 BSA) and the other (n=21) TID HC (10-15mg/m2 BSA). After one year, bone maturation ratio was kept stable in the PD group (from 1.20 to 1.14), whereas a slight increase was seen in the HC group (from 1.21 to 1.29). Growth velocity (SDS) was preserved in the PD group (from 1.2 to 1.2 in all; 0.79 to1.13 in pre-pubertals), whereas a slight increase occurred in the pre-pubertal HC-treated patients (from 1.1 to 1.9); height SDS for BA increased significantly in the PD group. Thus, patients with 21OHD treated for one year with a single morning dose of PD appear to achieve a better clinical and hormonal control than those on TID HC, permitting a reduction of the replacement dose. The current PD schedule used by our group (1.5-3mg/m2 BSA/day) suggests a higher HC:PD bioequivalence ratio of 6-8:1.
Keywords: 21-Hydroxylase deficiency; Prednisolone; Hydrocortisone; Growth; Androgens
RESUMO
Esquemas de reposição com hidrocortisona (HC) para a D21OH são, em geral, insatisfatórios e parcialmente eficientes quanto ao crescimento. Não aderência é comum já que a meia-vida da HC requer administração de 3 doses diárias. Mesmo várias doses não reproduzem a cronobiologia do cortisol e podem prejudicar os ritmos mediados pelo hipotálamo. Como glicocorticóides sintéticos podem melhorar o controle clínico, avaliamos os possíveis benefícios da terapia por um ano com fosfato de prednisolona (PD) em dose única matinal em 44 pacientes com D21OHD randomizados em 2 grupos: um (n=23) recebeu PD (2,4-3,5mg/m2SC) e o outro (n=21), HC 3 vezes ao dia (10-15mg/m2SC). Após um ano, a taxa de maturação óssea manteve-se estável no grupo PD (de 1,20 para 1,14), tendo aumentado discretamente (de 1,21 para 1,29) no grupo HC. A velocidade de crescimento (em SDS) foi preservada no grupo PD (de 1,2 para 1,2 no total de pacientes; 0,79 para 1,13 nos pré-púberes), enquanto discreto aumento ocorreu nos pré-púberes sob HC (1,1 to 1,9); a estatura para a IO (em SDS) elevou-se após um ano do uso de PD. Assim, pacientes com D21OH tratados por um ano com dose única matinal de PD parecem alcançar melhor controle clínico e hormonal do que aqueles usando 3 doses diárias de HC, permitindo inclusive a redução da dose. Nosso esquema atual de reposição com PD (1,5-3mg/m2SC/dia), sugere uma relação de bioequivalência HC:PD de 6-8:1.
Descritores: Deficiência de 21-hidroxilase; Prednisolona; Hidrocortisona; Crescimento; Andrógenos
INTRODUCTION OF GLUCO (GC) and mineralocorticoid replacement has significantly improved lifespan and the quality of life of patients with 21-hydroxylase deficiency (21OHD), but current therapeutic regimens are not completely satisfactory. Results regarding final height are generally disappointing, with losses consistently exceeding 1.0 SD (1). Additional problems as menstrual irregularities, infertility and psychosocial disturbances are also common (2-4).
The ultimate goal in treating 21OHD is to reach a balance between adequate GC supply and proper control of hyperandrogenism, in order to avoid acceleration of bone maturation and attain the predicted final height. However, handling GC replacement to achieve a suitable control of the disease is often difficult: when higher doses are occasionally necessary to reduce androgen excess, they may produce substantial side effects, especially impairment of linear growth and development; when dosage is dropped to prevent growth complications, they hardly control hirsutism and virilization, because ACTH suppression may not be effective. Thus, conventional medical therapy for 21OHD needs a constant fine-tuning to prevent any damage caused by either hyperandrogenism or hypercortisolism.
Among the several drugs and schedules proposed, hydrocortisone (HC) has often been preferred because, as the physiologic corticosteroid, it is deemed harmless regarding growth effects (5,6). Nevertheless, due to its short half-life and need for multiple daily intakes, HC fails in maintaining a good control of the disease, permitting an occasional escape of adrenal androgens (7-9). While total daily HC doses have been gradually decreased with time to fit actual cortisol production rates, it seems virtually impossible to reproduce its physiological circadian rhythm and pulsatile secretion. The replacement HC doses originally recommended in the 50's, ranged from 20 to 30mg/m2 BSA, a time when side effects due to overtreatment were common. It has been demonstrated that impairment in the final height can be avoided when smaller doses are used; at present, acceptable HC doses range from 10 to 15 mg/m2 BSA, in agreement with current data on cortisol secretion rate of 8-10mg/24hs (10,11).
When GCs other than HC are used, the bioequivalence dose ratio is based upon anti-inflammatory potency, because other clinical equivalence tables are not yet available. Thus, replacement therapy with synthetic GCs frequently uses improper doses, but even "physiologic doses" may impair growth velocity and restrict final height (12). Several mechanisms may be involved in this scenario: suppression of pituitary GH secretion, reduction of tissue sensitivity to GH, inhibition of IGF-1 bioactivity and decreased collagen synthesis (13).
Reliable results using small HC doses and mineralocorticoid replacement with fludrocortisone have been recently reported, when combined with anti-androgens (flutamide) and aromatase inhibitors (testolactone) (14). However, this complex and expensive multi-drug scheme is demanding for routine use, especially in third-world countries.
As an alternative option to treat 21OHD, long-acting synthetic GC analogues may suppress ACTH more efficiently (7,15,16). Prednisolone (PD) has a molecular structure that resembles that of cortisol, with the C1-C2 double bond determining a longer half-life and possibly permitting single daily dose administration. In addition, its convenient commercially available formulation (homogeneous oral solution) permits fine therapeutic adjustments.
In the present study, we have extended our previous short-term observations (17) for one year in order to evaluate potential clinical benefits of PD therapy in patients with 21OHD.
PATIENTS AND METHOD
Forty-four patients previously diagnosed as having the salt-losing (SL; n=27) or simple-virilizing (SV; n=17) forms of 21OHD were randomly assigned to two groups, stratified according to sex, age, and pubertal status, in order to assess the clinical benefits of a one-year treatment period with a single morning dose of PD, as compared to TID HC for GC replacement therapy. Group 1 patients (n=23) included 16F/7M, 1.6 to 20y of age (median: 9.4y), 10 pre-pubertal (B1 and G1, according to Tanners classification), 8 pubertal (B2-B4 and G2-G4), and 5 post-pubertal (adults, B5 and G5). Group 2 patients (n=21), encompassed 18F/3M, 1.2 to 21y of age (median: 8.3y), 11 pre-pubertal, 5 pubertal and 5 post-pubertal.
Group 1 received PD phosphate (Prelone®, 3mg/ml oral solution, Asta Medica, Brazil), initially at a dose of 2.4-3.75mg/m2 BSA once a day in the morning (7:00-8:00hs), whereas group 2 patients were maintained on oral HC acetate TID, 10-15mg/m2 BSA (half of the dose administered at 7:00-8:00hs, and 1/4 at 12:00-13:00hs and at 20:00-21:00hs). The initial HC to PD bioequivalence dose ratio employed was 4:1, as previously reported (18,19). All patients also received fludrocortisone acetate tablets, 0.1 mg per day in the morning, as mineralocorticoid replacement.
Patients were seen every 3mo by one of the authors (MCFC) at the Adrenal Outpatient Clinic from the Division of Endocrinology of UNIFESP, and at the Pediatric Endocrine Service, Hospital Darcy Vargas, São Paulo, SP, where clinical parameters (weight, height, growth velocity [GV], and pubertal stage) and serum levels of testosterone (T), androstenedione (A), and 17OH-progesterone (17OHP) were evaluated to monitor therapy. Dose adjustments were made whenever necessary, as judged by clinical and biochemical evaluation. Left wrist X-ray for bone age (BA) was obtained before and at the end of the one-year period. BA was estimated by the Greulich and Pyle method (20), and the BA/CA (chronological age) ratio was calculated.
The study protocol was approved by the Committees on Ethical Research from both institutions and informed written consents were obtained from all patients and/or their parents or legal guardians.
Standard deviation scores (SDS) for weight, height and GV for chronological age were determined according to Tanners standards (21). Height SDS for BA was also calculated.
All 10 post-pubertal ("adult") patients were excluded from the height, GV and BA analysis, because final stature and/or adult BA had been reached prior or soon after admittance to the study protocol. Another two female patients (7.4 and 9.7y), both from group 1, who had been on leuprolide acetate for true precocious puberty, were also excluded from the height and GV analysis.
All steroid determinations were performed in duplicate: T was determined by a routine RIE (22), whereas commercially available kits (Diagnostic Systems Laboratories-DSL, lnc, Welter, Tx) were used for A and 17OHP, with intra- and inter-assay precisions of 4.5% and 7%, and 8% and 8%, respectively.
Parametric and non-parametric statistics were used according to the nature of the variable. Mean±SD and median and range were used in the text and tables to represent central measurements and dispersion values for each parameter. Paired and unpaired t tests were used to compare initial and final (one-year) values and values between groups.
The data bank was statistically dealt with using the Graph Pad program, version 3.0 (PRISM TM). Statistic significance was set at the p<0.05 level.
RESULTS
Clinical and biochemical data at the beginning and at the end of the one-year treatment period are shown in tables 1 and 2, respectively.
Clinical data
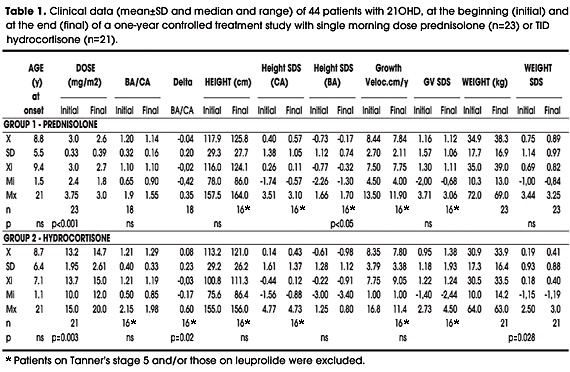
Patients in the PD group were slightly taller than those in the HC group, but the initial height SDS for CA was not statistically different between groups (table 1). Both the slight decrease in the PD group (0.37 to 0.11) and the slight increase in the HC group (-0.64 to 0.01) moved the median height SDS for CA towards zero at the end of the one. However, the absolute change (DBA/CA) after one year differed significantly between both groups (p<0.02) (figure 1), even when pubertal patients were excluded from analysis.
When height is corrected for BA, a significant increase in height SDS for BA is observed in the PD group (-0.73 to -0.17; p<0.05) at the end of one year, whereas it did not change in the HC group (-0.61 to -0.98; NS); although initial values were similar, final values were significantly lower in the HC group (-0.98 vs -0.17, respectively; p<0.02) (figure 2).
Growth velocity SDS for CA was similar in both groups and did not change significantly after one year (1.16 to 1.12 and 0.95 to 1.38), even when pre-pubertal patients were analyzed separately (0.79 to 1.13 and 1.13 to 1.92, respectively for PD and HC); a large individual variability was observed.
Seven of 10 patients (70%) from the PD group and 6 of 11 (54.5%) from the HC group continued on their Tanner's stage 1 pubertal status at the end of the one-year period.
Patients in the PD group were slightly heavier than those in the HC group; at the end of one year an average weight gain of 12% was observed for both groups. The initial weight SDS was not statistically different between the two groups; after one year it increased significantly in the HC group, but did not change in the PD group (table 1).
The average daily dose of PD at the onset of the study [3.0±0.3 (2.4-3.75) mg/m2 BSA] could be gradually diminished to 2.6±0.4 (1.8-3.0) mg/m2 BSA, a 15% reduction over one year. Conversely, the average HC daily dose was increased from 13.2±1.9 (10-15) to 14.9±2.6 (12-20) mg/m2 BSA, a 12% increase over the same period.
Hormonal data
Serum steroid levels at the beginning and after one year of treatment with PD or HC are shown in table 2. To avoid the influence of pubertal gonadal production, pre-pubertal and pubertal plus post-pubertal patients from both the PD and HC groups were analyzed separately. Although generally lower in the PD group, both initial and final values from all 3 steroids were not significantly different between groups. After one year of therapy serum levels decreased in both the pre- and pubertal PD subgroups (significantly for all, except A and 17OHP in pre-pubertal); a similar (but NS) pattern occurred with the HC subgroups.
Figure 3 discloses the average percent change of serum steroids after one year for the whole PD and HC groups, whereas figure 4 shows the individual final serum levels of T, A and 17OHP for both the HC and the PD groups, according to their pubertal status. Overall, a larger proportion of patients from the PD group were considered in good control (23), according to the hormonal cutoffs depicted in the figure.
No evidences of GC excess were observed during the treatment period, except for 2 female patients who gained distinctive weight and disclosed some facial plethora and mild hirsutism during the initial 3mo-course of PD. Symptoms relieved after appropriate dose reduction. Menstrual cycle abnormalities were present in 6 of 8 post-pubertal patients: 2 from the HC group remained primary amenorrheic and 4 were oligomenorrheic (2 from each group). The other 2, both from the PD group, had regular cycles.
DISCUSSION
21OHD is a peculiar endocrine disease in that clinical manifestations result from both a hormonal deficiency (hypocortisolism) and a hormonal excess (hyperandrogenism), consequences of a specific steroid biosynthetic deficiency in the adrenal cortex. Lifelong therapeutic control of both conditions is challenging and implies constant monitoring to yield adequate outcomes (24). In addition, poor adherence is common and prompts the clinician to seek for simpler treatment alternatives to suit patient's convenience and lifestyle.
Although considered the major GC, HC may not be appropriate to treat 21OHD. As suggested by a kinetic study of 17OHP, both the metabolism and clearance rates of HC were faster in patients than in controls (9). Also, questions regarding formulation of commercially available HC suspensions cast additional doubts on its bioavailability (25).
These points were also stressed in a recent consensus statement, in which oral HC suspension was not recommended for treatment of 21OHD (19).
In a previous short-term study, we observed that serum levels of adrenal steroids were better controlled on PD than on HC schemes (17). In the present study, we have extended that observation for one year in order to evaluate potential clinical benefits.
Regardless of previous GC treatment, the patients' heights at the onset of this study were adequate for chronological age, but sub-optimal when adjusted for bone age. The disproportionate increase in height SDS observed throughout the one-year controlled treatment period in the pre-pubertal patients who were on HC may reflect persistently mild hyperandrogenism. As a result, the target height potential was evidently limited in these patients, at the end of the observation period. On the other hand, PD treatment was able to hold excessive skeletal maturation and to improve the ultimate predicted height, as reflected by some gain in the height SDS for bone age in this group.
Absolute change of the BA/CA ratio was higher in the HC as compared to the PD group, demonstrating that HC is less efficient in restraining skeletal maturation. These results provide additional evidence that even on single morning dosages, PD is able to control bone maturation more efficiently than TID HC. Nevertheless, preservation of adequate growth rate for age in both groups was accompanied by a disproportionate advance in bone maturation, specially in pre-pubertal patients on HC. Moreover, considerable individual increases were observed in these patients, but not in those on PD. These data are in contrast with the widespread concern of a growth suppressive effect associated with the use of synthetic GCs (4,5). The potential for growth suppression linked to long-acting synthetic GC seems rather associated with the dose employed than with the GC itself. For instance, dexamethasone - which is usually used in adult patients with 21OHD -, proved adequate also in pre-pubertal patients provided a more physiologic bioequivalence ratio (DEX:HC 1:70) is respected (15). In our study, the growth pattern observed in the PD group is at least comparable to those previously reported on HC (26).
Since clinical signs may not be particularly evident at short-term intervals, therapeutic monitoring of 21OHD is based mainly on the concentration of serum steroids. However, it is precarious to ascertain daily production rates based on single morning samples because of their substantial variation throughout the day. 17OHP, in particular, is vulnerable for follow-up, due to its enormous circadian variations (24). Even so, significant decreases in all measured adrenal steroids were observed after one year on PD both in pre - and pubertal patients, a fact not evident for HC. After one year of treatment, appropriate serum testosterone and androstenedione values were seen in a much higher percentage of patients on PD than on HC. Again, adequate androgen control could be achieved with a once-a-day PD administration, raising an argument against the consensus statement recommendation of BID administration (19).
The best time of the day for GC intake is an additional matter of controversy in replacement therapy for 21OHD (16,27). Charmandari et al (28) observed different metabolic clearance rates for HC associated with the time of the drug intake: a 26% decrease was seen in the evening, as compared to the morning intake. The prolonged half-life at evening therapeutic schedules may have potential deleterious implications. We have previously shown that adrenal steroids were equally well controlled with either a morning or an evening dose of PD (17). However, when the non-physiologic evening schedule was employed, there were evident changes in the chronobiologic hormonal profile (a 4h-phase delay) and a higher prevalence of side effects (17).
Our results confirm a high prevalence of menstrual abnormalities in patients with 21OHD (3,29). Although a clear explanation is not available, chronic progesterone and/or androgen excess may play important roles. Because most Tanner's stage 5 patients had disordered cycles, an oral estrogen prescription should be added to their treatment.
Weight gain was observed in three children when starting on PD, but was corrected in two after dose adjustment. In the third one, personal and familial conflicts may have been important in changing her eating habits. A question of individual sensitivity and/or inadequate PD to HC bioequivalence can be invoked. Notwithstanding, PD was devoid of any significant side effects after one year of continuous treatment. To reinforce the inadequacy of the reported PD to HC bioequivalence ratio, clinical and biochemical monitoring of our patients prompted us to gradually decrease PD while increasing HC dosages along the year, resulting in an estimated HC to PD bioequivalence ratio of 6-8:1 after one year of treatment. Even a higher equivalence ratio (15:1) was recently reported by Punthakee et al (30). At present, we have successfully reduced the replacement PD dose range in our 21OHD patients to 1.5-3.0 mg/m2 BSA/day.
Some additional information obtained in this study is worth to mention: parents refer that dose skipping in TID HC was frequent, whereas the once-a-day PD schedule favored adherence, facilitating the household routine. When cost-benefit is concerned, the monthly cost to treat a 1m2 BSA child with 21OHD is of approximately US$ 1.8 with PD vs. US$ 6.0 with HC (as per March 2004 in Brazil).
Therefore, several pieces of evidence favor the use of PD for the treatment of 21OHD. Of all synthetic GC, PD has the most structurally closer molecule to HC. As the biologic active form of the commonly used prednisone, it bypass liver activation, which could be a problem when the physiologic hepatic inability of childhood or adolescence (11bHSD1 deficiency) is considered (31,32). A single daily PD dose has been proven as efficient and adequate as TID HC for clinical and biochemical control of 21OHD patients. Once-a-day intake of PD is more convenient, reliable, and improves treatment compliance, in addition to its cost-effectiveness. Also, PD is commercially available and its liquid formulation is useful in fine-tuning and tailoring patient's dosage, as opposed to HC tablets. Most patients kept on PD have maintained expected good results, similar or better than with HC.
In conclusion, when the convenience of a single daily dose, compliance and cost-effectiveness are considered, PD proves superior to HC for the continuing medical treatment of 21OHD. However, long-term follow-up is still necessary to demonstrate individual clinical benefits upon final stature and fertility.
ACKNOWLEDGMENTS
The authors wish to thank Dr. leda Verreschi for helpful suggestions and comments.
Recebido em 20/06/04
Aceito em 03/07/04
-
11. Eugster EA, DiMeglio LA, Wright JC, Freidenberg GR, Seshadri R, Pescovitz OH. Height outcome in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency: a meta-analysis. J Pediatr 2001;138:26-32
-
22. Jaaskelainen J, Tiitinen A, Voutilainen R. Sexual function and fertility in adult females with congenital adrenal hyperplasia. Horm Res 2001;56:73-80
-
33. Holmes-Walker DJ, Conway GS, Honour JW, Rumsby G, Jacobs HS. Menstrual disturbance and hypersecretion of progesterone in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol 1995;43:291-6
-
44. Berenbaum SA. Effects on early androgens on sex-typed activities and interests in adolescents with congenital adrenal hyperplasia. Horm Behav 1999;35:102-10
-
55. Merke DP, Cutler GB. New ideas for medical treatment of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2001;30:121-35
- 6. Styne DM, Richard GE, Bell JJ. Correlation of glucocorticoid therapy with stature. In: Lee PA, Plotnik LP, Kowarski AA, eds. Congenital adrenal hyperplasia. Baltimore: University Park Press, 1977. p.247-64
-
77. Husseman CA, Varma MM, Blizzard RM, Johason A. Treatment of congenital virilizing hyperplasia patients with single and multiple daily doses of prednisone. J Pediatr 1977;90:538-42
-
88. Zipf WB, Bacon GE, Kelch RP. Hormonal and clinical responses to prednisone treatment in adolescents with congenital adrenal hyperplasia. Horm Res 1980;12:206-17
-
99. Groschl M, Rauh M, Dorr H. Cortisol and 17-hydroxyprogesterone kinetics in saliva after oral administration of hydrocortisone in children and young adolescents with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2002;87:1200-4
-
1010. Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990;117:892-6
-
1111. Kerrigan JR, Velchius JD, Leyo SA, lranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal puberty males by deconvolution analysis. J Clin Endocrinol Metab 1993;76:1505-10
-
1212. Muhlendahl KE, Weber B, Muller-Hess R, Korth-Schutz S, Helge H. Glucocorticoids and growth hormone secretion under physiological conditions and in states of steroids excess. Eur J Pediatr 1978;128:113-21
-
1313. Tonshoff B, Mehls O. Interactions between glucocorticoids and the somatotropic axis. Acta Paediatr (Suppl) 1996;417:72-5
-
1414. Merke DP, Keil MF, Jones JV, Fields J, Hill S, Cutler G. Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2000;85:1114-20
-
1515. Rivkees SA, Crawford JD. Dexamethasone treatment of virilizing congenital adrenal hyperplasia: the ability to achieve normal growth. Pediatrics 2000;106:767-73
-
1616. Hayek A, Crawford JD, Bode HH. Single dose of dexamethasone in treatment of congenital adrenal hyperplasia. Metabolism 1971;20:897-90
-
1717. Fernandes-Caldato MC, Fernandes VT, Kater CE. A chronobiological evaluation of adrenal androgens in response to prednisolone or hydrocortisone in morning and evening schemes for treatment of classic 21-hydroxylase deficiency. J Clin Endocrinol Metab (Submitted 2004)
-
1818. Corticosteroids - Glucocorticoids effects. Drug information for the health care professional. USP.Dl 2002; http://micromedex.com/products/uspdi/v1/
-
1919. Joint LWPES/ESPE CAH Working group. Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab 2002;87:4048-53
- 20. Greulich WW, Pyle Sl. Radiographic atlas of skeletal development of the hands and wrists. Standford: Standford University Press;1959
-
2121. Tanner JM, Whitehouse RH, Takaishi M. Standards from birth to maturity for height, weight, height velocity and weight velocity: British children, 1965. Part II. Arch Dis Child 1966;41:613-35
-
2222. Vieira JGH, Nishida SK, Lombardi MT, Noguti KO. Evaluation of a radioimmunoassay for determination of serum testosterone using a monoclonal antibody (portug). J Bras Patol 1996;32:56-63
-
2323. Young MC, Robinson JA, Read GF, Riad-Fahmy D, Hughes IA. 17OH-progesterone rhythms in congenital adrenal hyperplasia. Arch Dis Child 1988;63:617-23
-
2424. Hughes IA, Dyas J, Robinson J, Walker RF, Fahmy DR. Monitoring treatment in congenital adrenal hyperplasia: use of serial measurements of 17OH-progesterone in plasma, capillary blood, and saliva. Ann NY Acad Sci 1985;458:193-202
-
2525. Merke, DP, Cho D, Calis KA, et al. Hydrocortisone suspension and hydrocortisone tables are not bioequivalent in the treatment of children with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2001;86:441-5
-
2626. Merke DP, Stefan R, Bornstein MD, Avila NA, Chrousos GP. Future directions in the study and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. NIH Conference. Ann lntern Med 2002;136:320-34
-
2727. Nichols T, Nugent CA, Tyler FH. Diurnal variation in suppression of adrenal function by glucocorticoids. J Clin Endocrinol Metab 1965;25:343-8
-
2828. Charmandari E, Johnston A, Brook CGD, Hindmarsh PC. Bioavailability of oral hydrocortisone in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Endocrinol 2001;169:65-70
-
2929. Richards GE, Grumbach MM, Kaplan SL, Conte FA. The effect of long acting glucocorticoids on menstrual abnormalities in patients with virilizing congenital adrenal hyperplasia. J Clin Endocrinol Metab 1978;47:1208-15
-
3030. Punthakee Z, Legault L, Polychronakos C. Prednisolone in the treatment of adrenal insufficiency: a re-evaluation of relative potency. J Pediatr 2003;143:402-5.
-
3131. Whorwood CB, Warne GL. A possible defect in the inter-correlation in prepubertal patients with congenital adrenal hyperplasia receiving cortisone acetate therapy. J Steroid Biochem Molec Biol 1991;39(4A):461-70
-
3232. Jinno K, Sabura N, Nomura S, Fujitaka M, Ueda K, Kihra M. Failure of cortisone acetate therapy in 21-hydroxylase deficiency in early infancy. Pediat Intern 2001:43:478-82
Correspondence to
Publication Dates
-
Publication in this collection
07 Mar 2005 -
Date of issue
Oct 2004
History
-
Received
20 June 2004 -
Accepted
03 July 2004