Resumos
Glicocorticóides são amplamente utilizados na prática clínica para o controle da atividade de doenças auto-imunes, inflamatórias, alérgicas e outras entidades nosológicas. Doses terapêuticas de glicocorticóides são muita vezes administradas inapropriadamente e isto é um problema particular, pois a terapia crônica tem muitos efeitos colaterais que se estendem desde a supressão do eixo hipotálamo-hipofisário-adrenal e síndrome de Cushing até infecções e alterações do status mental. Fatores que influenciam tanto nos efeitos adversos quanto nos terapêuticos dos glicocorticóides incluem propriedades farmacocinéticas do glicocorticóide, dose diária, diferenças individuais no metabolismo esteróide e duração do tratamento. Quando utilizados para o controle da atividade destas doenças, quatro aspectos da retirada de glicocorticóide merecem atenção especial. Primeiro, a doença tratada pelo esteróide pode recorrer. Segundo, o eixo hipotálamo- hipófise-adrenal pode permanecer suprimido por um longo período. Terceiro, muitas vezes desenvolve-se dependência psicológica a esses hormônios. Quarto, uma síndrome de retirada inespecífica pode desenvolver mesmo enquanto os pacientes estão recebendo doses de reposição fisiológica de glicocorticóides. A gravidade da síndrome de retirada depende da fase e o grau de dependência e inclui sintomas tais como anorexia, náusea, vômitos, perda de peso, fadiga, mialgias, artralgias, cefaléia, dor abdominal, letargia, hipotensão postural, febre e descamação da pele.
Glicocorticóide; Síndrome de retirada; Esteróide
Glucocorticoids are widely used in clinical practice to control the activity of autoimmune, inflammatory, allergic diseases and other nosological entities. Therapeutic doses of glucocorticoids are often administered inappropriately and it is a particular problem because chronic therapy has many side effects, ranging from suppression of the hypothalamic-pituitary-adrenal axis and Cushing's syndrome to infections and changes in mental status. Factors influencing both the therapeutic and adverse effects of glucocorticoids include the pharmacokinetic properties of the glucocorticoid, daily dosage, individual differences in steroid metabolism and the duration of treatment. When used to control the activity of these diseases, four aspects of glucocorticoid withdrawal deserve special attention. First, the illness treated by steroids may relapse. Second, the hypothalamic-pituitary-adrenal axis may remain suppressed for a long time. Third, psychological dependence to these hormones often develops. Fourth, a nonspecific withdrawal syndrome may develop even while patients are receiving physiological replacement doses of glucocorticoids. The severity of the withdrawal syndrome depends on the phase and degree of dependence and includes many symptoms as anorexia, nausea, emesis, weight loss, fatigue, myalgias, arthralgias, headache, abdominal pain, lethargy, postural hypotension, fever, and skin desquamation.
Glucocorticoid; Withdrawal syndrome; Steroid
REVISÃO
Síndrome de Cushing exógena e retirada de glicocorticóides
Exogenous Cushing's syndrome and glucocorticoid withdrawal
Daniella J.P.C. Romanholi; Luiz Roberto Salgado
Unidade de Neuroendocrinologia da Disciplina de Endocrinologia do Hospital das Clínicas, Universidade de São Paulo - HCFMUSP, São Paulo, SP
Endereço para correspondência Endereço para correspondência: Luiz Roberto Salgado Av. Brigadeiro Luiz Antônio 4258 01402-002 São Paulo, SP Fax: (11) 3885-6719 E-mail: salga@uol.com.br
RESUMO
Glicocorticóides são amplamente utilizados na prática clínica para o controle da atividade de doenças auto-imunes, inflamatórias, alérgicas e outras entidades nosológicas. Doses terapêuticas de glicocorticóides são muita vezes administradas inapropriadamente e isto é um problema particular, pois a terapia crônica tem muitos efeitos colaterais que se estendem desde a supressão do eixo hipotálamohipofisárioadrenal e síndrome de Cushing até infecções e alterações do status mental. Fatores que influenciam tanto nos efeitos adversos quanto nos terapêuticos dos glicocorticóides incluem propriedades farmacocinéticas do glicocorticóide, dose diária, diferenças individuais no metabolismo esteróide e duração do tratamento. Quando utilizados para o controle da atividade destas doenças, quatro aspectos da retirada de glicocorticóide merecem atenção especial. Primeiro, a doença tratada pelo esteróide pode recorrer. Segundo, o eixo hipotálamo hipófiseadrenal pode permanecer suprimido por um longo período. Terceiro, muitas vezes desenvolve-se dependência psicológica a esses hormônios. Quarto, uma síndrome de retirada inespecífica pode desenvolver mesmo enquanto os pacientes estão recebendo doses de reposição fisiológica de glicocorticóides. A gravidade da síndrome de retirada depende da fase e o grau de dependência e inclui sintomas tais como anorexia, náusea, vômitos, perda de peso, fadiga, mialgias, artralgias, cefaléia, dor abdominal, letargia, hipotensão postural, febre e descamação da pele.
Descritores: Glicocorticóide; Síndrome de retirada; Esteróide
ABSTRACT
Glucocorticoids are widely used in clinical practice to control the activity of autoimmune, inflammatory, allergic diseases and other nosological entities. Therapeutic doses of glucocorticoids are often administered inappropriately and it is a particular problem because chronic therapy has many side effects, ranging from suppression of the hypothalamic-pituitary-adrenal axis and Cushing's syndrome to infections and changes in mental status. Factors influencing both the therapeutic and adverse effects of glucocorticoids include the pharmacokinetic properties of the glucocorticoid, daily dosage, individual differences in steroid metabolism and the duration of treatment. When used to control the activity of these diseases, four aspects of glucocorticoid withdrawal deserve special attention. First, the illness treated by steroids may relapse. Second, the hypothalamic-pituitary-adrenal axis may remain suppressed for a long time. Third, psychological dependence to these hormones often develops. Fourth, a nonspecific withdrawal syndrome may develop even while patients are receiving physiological replacement doses of glucocorticoids. The severity of the withdrawal syndrome depends on the phase and degree of dependence and includes many symptoms as anorexia, nausea, emesis, weight loss, fatigue, myalgias, arthralgias, headache, abdominal pain, lethargy, postural hypotension, fever, and skin desquamation.
Keywords: Glucocorticoid; Withdrawal syndrome; Steroid
A SÍNDROME DE CUSHING (SC) Pode ser dividida em dois grandes grupos segundo a sua etiologia: ACTH(corticotrofina)-dependente e ACTH-independente.
A SC exógena ou iatrogênica é considerada a causa mais comum devido à vasta utilização de glicocorticóides (GC) sintéticos pela população e se encontra dentre as causas ACTH-independentes. É descrita a SC ACTH-dependente iatrogênica que está relacionada à administração de ACTH exógeno, o que representa menos de 1% de causas de SC de origem factícia (2).
O acetato de megestrol e seu agente relacionado medroxiprogesterona são progestógenos utilizados no manejo da caquexia da SIDA (síndrome da imunodeficiência adquirida) e no tratamento de neoplasias da mama e próstata. Embora não sejam considerados GC, podem apresentar efeito GC quando utilizados em altas doses e inibir o eixo hipotálamo-hipófise-adrenal (HHA) causando SC e insuficiência adrenal quando interrompidos de maneira abrupta (3).
FISIOLOGIA
Os glicocorticóides são sintetizados a partir do colesterol e produzidos na zona fasciculada da supra-renal sob controle do eixo HH. A síntese de mineralocorticóides ocorre na zona glomerulosa por estímulo do sistema reninaangiotensinaaldosterona (SRAA), hipovolemia e hipercalemia.
A produção estimada de cortisol é de aproximadamente 5 mg/m2 a 10 mg/m2 por dia (o equivalente a 2030 mg/dia de hidrocortisona, 57 mg/dia de prednisona e 2537,5 mg de acetato de cortisona) (4).
A síntese de cortisol pode aumentar cerca de 5 a 10 vezes em condições de estresse, chegando a um nível máximo de aproximadamente 100 mg/m2/dia (4).
FISIOPATOLOGIA
Glicocorticóides sintéticos e naturais podem ser utilizados em diversas doenças endócrinas ou não endócrinas. Na área de endocrinologia, os GC são utilizados em testes para se estabelecer diagnóstico e confirmar a etiologia da SC, para o tratamento de insuficiência adrenal e na hiperplasia adrenal congênita.
Os GC também são aplicados na prática clínica em doses farmacológicas no tratamento de pacientes portadores de patologias inflamatórias, alérgicas, imunológicas, reumatológicas etc. Infelizmente, os GC são utilizados de maneira empírica e inapropriada sem que a eficácia clínica ou o mecanismo de ação sejam rigorosamente estabelecidos (5).
Diversos fatores influenciam nos efeitos terapêuticos e adversos dos GC, dentre eles: propriedades farmacocinéticas do GC em uso, dose diária, fracionamento de doses, duração do tratamento e diferenças individuais no metabolismo esteróide.
GLICOCORTICÓIDES SINTÉTICOS
Estrutura dos esteróides
Modificações químicas nos esteróides naturais levam às alterações essenciais para a atividade biológica dos diferentes GC sintéticos. A configuração delta4, 3ceto11beta, 17alfa 21triidroxil está presente em todos os esteróides naturais e sintéticos (figura 1 e figura 2).
Prednisona, o GC mais utilizado na prática clinica, é a prednisolona com um radical 11ceto no local 11betahidroxi. É uma molécula inativa que precisa ser convertida em prednisolona no fígado para se tornar ativa (6). Outras modificações na molécula da prednisolona também produzem outros GC com potências biológicas diferentes. A adição do grupo 6alfametil na molécula da prednisolona origina a metil prednisolona que é 5 vezes mais potente que o cortisol (7).
A adição de um átomo de flúor na posição 9alfa de hidrocortisona produz a fludrocortisona, que possui uma atividade glicocorticóide 12 vezes maior que o cortisol e uma atividade mineralocorticóide 125 vezes maior que o cortisol (8).
A adição do grupo 16alfametil na molécula delta1fludrocortisona produz a dexametasona, que apresenta uma potência glicocorticóide 30 vezes maior que a hidrocortisona, porém com atividade mineralocorticóide desprezível (9).
A betametasona é uma dexametasona com um radical 16betametil e não alfametil, possuindo atividade GC semelhante à dexametasona e normalmente é utilizada sob a forma de aerosol.
A triamcinolona é uma dexametasona com um grupo 16alfahidroxil e não metil e geralmente é utilizada em terapias tópicas.
Deflazacort é um análogo da prednisolona oxazilínico inativo que é convertido em um metabólito 21desacetil, com efeito antiinflamatório semelhante à prednisolona, porém com menor efeito no metabolismo de carboidratos e ósseo (10,11).
Farmacocinética
O cortisol sérico circula ligado à CBG (cortisol binding globulin) e à albumina, e grande parte do cortisol ativo circula ligada aos eritrócitos (12).
Os GC sintéticos possuem uma afinidade muito baixa à CBG e aos eritrócitos; sendo assim, 2/3 ligam-se fracamente à albumina e 1/3 circula sob a forma livre (13-16). Esta característica possibilita que os GC sintéticos apresentem uma meia-vida mais longa que o cortisol. A meia-vida dos GC sintéticos varia para cada indivíduo de acordo com a idade (17) genética e a influência de outras drogas (tabela 1). Nos idosos, a meia-vida dos GC é mais prolongada.
Em contraste com a fraca afinidade pela CBG, os GC sintéticos apresentam elevada afinidade pelos seus receptores (tipo II). A afinidade da prednisolona e triamcinolona é 2 vezes maior que a do cortisol, enquanto que a metil-prednisolona chega a ser 11 vezes maior (12). A prednisona possui baixa afinidade pelos receptores de GC, conseqüentemente seu efeito glicocorticóide é desprezível sendo necessária sua conversão à prednisolona para sua ação, semelhante ao que ocorre com a cortisona, que deve ser convertida a cortisol pela 11betahidroxiesteróide desidrogenase (6).
Potência biológica e duração da ação
Tanto a afinidade do esteróide pelo receptor de GC quanto a sua farmacocinética não são preditores fiéis da potência biológica de cada GC. Essa é influenciada por outros fatores como: eficiência e taxa de absorção pela circulação sistêmica e o seu respectivo metabolismo. As características farmacocinéticas e de potência biológica dos diversos GC são descritas na tabela 1. Interações com outras drogas também podem ser observadas (tabela 2).
Os GC orais são absorvidos geralmente em 30 minutos (18); já os tópicos variam sua absorção de acordo com diversos fatores (região de aplicação, veículo utilizado, oclusão do local, idade do paciente).
A taxa de absorção dos GC parenterais varia bastante, assim como aqueles administrados por via intra-articular.
USO FARMACOLÓGICO DO GC
Glicocorticóides sistêmicos
A prescrição de um GC sistêmico pode estar relacionada a diversas situações (doenças crônicas e emergenciais). Quadros de emergência (choque anafilático, nefrite lúpica, choque séptico, síndrome enxerto X hospedeiro, broncoespasmo grave etc.) podem necessitar do uso de GC. Nesses casos, geralmente a administração é realizada por via parenteral, em altas doses e por poucos dias (pulsoterapia) até que a terapia primária estabelecida apresente eficácia.
A terapia crônica com GC é empregada em diferentes doenças anteriormente descritas e é nessa situação que o médico deve estar atento aos riscos da corticoterapia prolongada e abusiva. Assim, alguns critérios devem ser obedecidos ao iniciar uma terapia crônica com GC (tabela 3).
Efeitos adversos do GC sistêmicos
Muitos dos efeitos colaterais da corticoterapia podem ser confundidos com os achados da SC propriamente dita. A principal diferença entre a SC exógena e a endógena é que esta última apresenta maior ação mineralocorticóide, resultando em maior incidência de hipertensão arterial sistêmica (HAS), hipocalemia e sintomas de hiperadrogenismo (hirsutismo e sintomas de virilização). Além disso, alguns efeitos menos comuns tais como o pseudo-tumor cerebral, necrose óssea avascular, lipomatose epidural espinhal, catarata e glaucoma estão mais relacionados à SC exógena. (19)
Os GC de uso tópico ou intra-articular podem sofrer absorção sistêmica levando também ao aparecimento de efeitos colaterais e SC. Os fatores que aumentam a absorção sistêmica de GC intra-articular são injeções freqüentes e o uso de elevadas doses.
Alguns fatores podem influenciar na absorção sistêmica dos GC tópicos: uso de agentes de alta potência, áreas extensas de aplicação e a oclusão do local onde foi aplicado o GC. A atrofia cutânea, despigmentação local e o aparecimento de teleangectasias podem ser alguns efeitos locais do uso de GC tópico.
São descritos na literatura casos de SC em crianças que receberam injeções de GC em quelóides ou outras cicatrizes (queimadura) após um tempo relativamente prolongado, e acredita-se que a pouca vascularização desses tecidos cicatriciais possa ocasionar a absorção mais lenta do GC (20,21).
Curiosamente, já foram descritos relatos de SC induzida pelo bloqueio do nervo occipital com triamcinolona (22) e crise de insuficiência adrenal em uma paciente que utilizou GC retal (23).
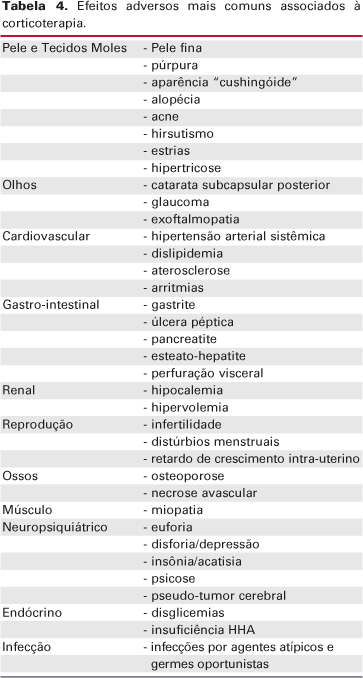
Um dado interessante é o fato de que a terapia combinada de GC (por qualquer via) associada a antifúngicos aumenta a incidência do desenvolvimento de SC. Isso porque qualquer agente que interfira com o sistema citocromo p450 altera o metabolismo do GC. Diversos casos de SC são descritos em pacientes com fibrose cística que normalmente recebem corticoterapia associada ao itraconazol e também em portadores de SIDA durante o uso de ritonavir associado à fluticasona (24-27). As complicações da corticoterapia serão descritas de acordo com o sistema em questão (tabela 4).
No intuito de minimizar ou diagnosticar precocemente essas complicações, algumas medidas são importantes durante o acompanhamento do paciente em corticoterapia (tabelas 5).
Pele e partes moles
O hipercortisolismo inibe a proliferação de fibroblastos levando à perda de colágeno e tecido conectivo. Os efeitos colaterais mais comuns na pele e tecidos moles dos pacientes em corticoterapia são a atrofia cutânea e o aparecimento de púrpuras. Essas geralmente atingem áreas expostas ao sol (dorso das mãos e ombros) e não são acompanhadas de edema palpável (28). Estrias e dificuldade na cicatrização também podem estar presentes.
Neoplasias de pele (carcinoma espinocelular e carcinoma basocelular) também são mais freqüentes nos usuários de GC (29,30).
O uso de GC tópicos pode levar ao surgimento de acne, alopécia, hipertricose e estrias (31).
Olhos
A catarata está associada ao uso prolongado de GC pela ligação covalente dos esteróides às proteínas do cristalino (32,33). Geralmente, diferencia-se da catarata senil por ser de localização posterior, bilateral, de desenvolvimento lento e que tende a se estabilizar com a redução da dose do GC. As crianças tendem a ser mais susceptível que os adultos. Alguns trabalhos sugerem que este efeito é dose e tempo dependente e que raramente aparece em tratamentos com menos de 1 ano de duração ou uso de doses de prednisona inferiores a 10 mg/d. Outros acreditam que não existe uma dose segura para se evitar esse efeito (34,35).
Os GC em colírios e mesmo por via oral aumentam a pressão intra-ocular levando ao surgimento de glaucoma de ângulo aberto (36-38). Acredita-se que, geralmente, esses indivíduos apresentam alguma predisposição genética (história familiar positiva para glaucoma). A coriorretinopatia é outro efeito colateral raro (39).
Doença cardiovascular e aterosclerose
O uso de doses suprafisiológicas de GC está associado ao aumento da incidência de infarto do miocárdio, acidente vascular cerebral e insuficiência cardíaca (40,41). O risco dessas complicações é proporcional à dose de GC utilizada e tende a ser maior quando há uso contínuo comparado ao uso intermitente.
GC também estão relacionados ao desenvolvimento da doença vascular periférica. As alterações descritas são mediadas pelos distúrbios metabólicos causados pelos GC, tais como resistência à insulina, hiperinsulinemia e aumento da síntese de VLDL-colesterol (42). Em trabalho recente, observou-se que a administração de ACTH por 3 semanas a indivíduos com dislipidemia em corticoterapia prolongada determinou a redução do LDL-colesterol e dos triglicérides e o aumento do HDL-colesterol. Esses dados reforçam a teoria de que o ACTH suprimido pela corticoterapia crônica seria uma das causas de alterações lipídicas nesses indivíduos (43).
Trato gastro-intestinal
Os GC induzem a produção de ácido clorídrico pela mucosa gástrica aumentando o risco de úlcera péptica, gastrite e sangramento gastro-intestinal. Em um estudo com 477 portadores de artrite reumatóide em corticoterapia prolongada, 7,5% apresentaram úlcera péptica (44). Este efeito é menor que o relacionado aos antiinflamatórios não hormonais (AINH), considerando-se um efeito sinérgico entre eles. O uso isolado de GC não requer profilaxia contra sangramento gastro-intestinal, mas seu uso em combinação com aspirina ou outros AINH é indicação para a necessidade de medicação preventiva.
Rins e sistema hemodinâmico
Os esteróides afetam o balanço hidro-eletrolítico e esse efeito é decorrente da ocupação dos receptores de mineralocorticóides (retenção de sódio, hipocalemia, alcalose metabólica e hipertensão arterial) ou via glicocorticóide (aumento da taxa de filtração glomerular pelo aumento do débito cardíaco ou efeito renal direto). A retenção hídrica não afeta indivíduos normais mas seria risco potencial para portadores de certo grau de insuficiência renal ou cardíaca.
A corticoterapia está relacionada ao surgimento de hipertensão arterial sistêmica, complicação de fisiopatologia não bem definida (45-47).
Sistema reprodutivo
Hipercortisolismo pode causar irregularidade menstrual e infertilidade tanto no homem como na mulher, provavelmente pela inibição da secreção das gonadotrofinas. Alem desse efeito central, em nível ovariano e testicular observa-se inibição da produção de estrógeno ou testosterona pelas células da granulosa e de Leydig, respectivamente (48). A corticoterapia na gestação está associada ao risco de fenda palatina.
Sistema músculo-esquelético
A fraqueza muscular é uma queixa rara dos usuários de GC e em geral é proximal, motora, de membros superiores e inferiores e cursa com níveis normais de enzimas musculares (CK). Vale ressaltar que altas doses de GC empregadas por períodos prolongados (após radioterapia do SNC) podem resultar em diminuição da força muscular (miopatia esteróide).
A osteonecrose é uma complicação rara e muito grave do hipercortisolismo principalmente endógeno. O local de acometimento mais comum é o fêmur proximal (49) e o risco é proporcional à dose de GC; no entanto, pode ocorrer após o uso de GC intra-articular ou mesmo em doses baixas por tempo prolongado (50,51).
Os GC inibem diretamente a formação óssea através da diminuição da proliferação celular, síntese de RNA, proteínas e colágeno (36,52,53). Além disso, estimulam a reabsorção óssea potencializando as ações do PTH e da 1,25-vitamina D no osso. Reduzem a absorção intestinal de cálcio e promovem fosfatúria e hipercalciúria. O balanço do cálcio negativo (e conseqüentemente o hiperparatireoidismo secundário) associado à diminuição da formação óssea (54) e o aumento de reabsorção óssea (55,56) resultam em aparecimento de osteoporose (57-60). Essa diminuição da densidade mineral óssea ocorre principalmente em osso trabecular.
Além dos efeitos diretos exercidos pelos GC na remodelação óssea, também se observa um efeito indireto, não menos importante devido a sua ação no eixo HHA e gonadal por bloqueio em nível hipofisário da resposta do LH ao LHRH, reduzindo os níveis de estrogênios e testosterona gonadais e androgênios adrenais.
A perda óssea parece estar acelerada nos primeiros seis meses de terapia, mas persiste mais lentamente após esse período (61). O risco de fratura diminui rapidamente após a suspensão do GC (62).
Em um ensaio clínico randomizado com pacientes portadores de artrite reumatóide, houve uma queda de 8,2% na densidade mineral óssea da coluna lombar após 20 semanas do uso de prednisona 50 mg/semana e melhora importante após 24 semanas de suspensão da corticoterapia (63).
A ocorrência de fraturas vertebrais nos usuários de corticóides que apresentavam densidade mineral óssea normal sugere um mecanismo adicional de fragilidade óssea no hipercortisolismo (46,64), salientando-se que fraturas podem ocorrer em 30 a 50% dos pacientes em corticoterapia crônica (61).
Crescimento
Os GC em excesso inibem o crescimento longitudinal nas crianças através do seu efeito direto no osso, mas também por interferirem na secreção de GH (growth hormone), na geração de IgF1, bem como sua ação na cartilagem de crescimento.
Tireóide
Hipercortisolismo pode inibir a síntese e a secreção de TSH e a resposta de TSH ao TRH.
Geralmente, os níveis de T4 total estão subnormais pela redução da TBG (thyroxine binding globulin), mantendo os níveis de T4 livre adequados.
Os GC diminuem a conversão de T4 em T3 e aumentam a síntese de T3 reverso. Apesar dessas alterações, manifestações de hipotireoidismo estão geralmente ausentes.
Tecido adiposo
Os GC aumentam a lipólise elevando dos níveis de glicerol e ácidos graxos livres. Apesar de lipolítico, o aumento na deposição de gordura visceral é característico do hipercortisolismo. Esse efeito paradoxal pode ser justificado pelo aumento do apetite associado aos esteróides e ao efeito lipogênico da hiperinsulinemia presente no hipercortisolismo com depósito de gordura na região central (na face, região cervical, tronco e abdome preservando as extremidades). Essa variabilidade de resposta do tecido adiposo deve-se à heterogeneidade da gordura corporal (66).
Sistema nervoso central
Muitos pacientes em uso de GC referem uma sensação de bem-estar que parece ser independente da melhora da doença de base. O desenvolvimento de uma sensação de euforia não é incomum nesses pacientes e alguns podem apresentar distúrbios psiquiátricos (67). História familiar de depressão ou alcoolismo aumenta o risco do surgimento desses distúrbios com o uso de GC (68).
Lítio costuma ser o fármaco de escolha para o manejo e a prevenção de psicopatias associadas ao uso de GC (69).
As psicoses podem surgir, mas geralmente nos indivíduos que utilizam doses superiores a 20 mg/d de prednisona por tempo prolongado (70). Hipoalbuminemia parecer ser um fator de risco para a psicose induzida pelos GC (71).
Em relação ao sono, existe tendência à redução da fase REM e incremento da fase II com freqüentes queixas de insônia (72).
Pseudo-tumor cerebral raramente tem sido descrito em usuários de GC e, curiosamente, corticoterapia em altas doses é utilizada para aliviar os sintomas dessa patologia auto-limitada (73).
Metabolismo dos carboidratos
Os GC podem interferir no metabolismo de glicose, pois inibem a captação periférica de glicose pelo músculo e tecido adiposo. Além disso, aumentam a gliconeogenêse hepática (37) e promovem um efeito permissivo às ações do glucagon e catecolaminas (36,37), contribuindo para o estado de hiperglicemia.
Os GC têm sido descritos como promotores da diferenciação e proliferação dos adipócitos em humanos. Esses adipócitos tendem a se acumular no tecido adiposo visceral, o que está associado ao quadro de resistência insulínica presente na maioria dos pacientes com hipercortisolismo (74).
O desenvolvimento do diabetes mellitus (DM) é muito raro em pacientes que apresentam tolerância à glicose (75) sem alterações.
O DM associado à corticoterapia é facilmente controlado com a diminuição da dose do GC e costuma desaparecer após a sua suspensão (76).
Sistema imunológico
Os GC aumentam o número de leucócitos polimorfonucleares que são liberados da medula óssea e diminuem sua aderência ao endotélio. Também reduzem o número de linfócitos, monócitos e eosinófilos (77).
Os GC possuem diversos efeitos deletérios tanto na imunidade inata quanto adquirida, aumentando a predisposição a processos infecciosos. Esses efeitos incluem a diminuição da produção de citocinas pró-inflamatórias (prostaglandinas e interleucinas) e da função fagocítica (78).
O risco do processo infeccioso varia de acordo com a doença de base, local de tratamento do paciente, uso de terapias concomitantes e o tipo de corticoterapia. É sabido que o risco do processo infeccioso pode ser significantemente diminuído com o uso de GC de curta ação (prednisona) administrado em dias alternados (79).
Infecções atípicas (Herpes zoster) ou com organismos oportunistas (pneumocistis carinii) são 40 vezes mais freqüentes em pacientes em corticoterapia (80). Quando associados a outras drogas imunossupressoras, também pode ocorrer a reativação de uma infecção latente. Alguns autores sugerem que pacientes com PPD positivo, raios-X de tórax sugestivo de tuberculose ou história familiar devem receber profilaxia contra tuberculose antes ou concomitante à corticoterapia.
Sistema endocrinológico
Os GC exógenos também atuam no eixo HHA, suprimindo a produção de CRH (corticotrophin releasing hormone) hipotalâmico e de ACTH hipofisário. A corticoterapia prolongada pode levar à diminuição acentuada da secreção de cortisol endógeno. O primeiro caso de insuficiência adrenal após o uso de GC exógeno foi descrito em 1961 (81).
O tempo necessário para a supressão do eixo HHA, assim como o desenvolvimento de SC exógena, varia em cada indivíduo e está relacionado com a dose, horário de administração, tipo e a duração da corticoterapia.
As manifestações clínicas da SC exógena são parcialmente semelhantes àquelas presentes na SC endógena, porém diferem desta em relação ao tempo de evolução. No hipercortisolismo endógeno o quadro clínico tende a ser gradual e de evolução mais lenta.
GLICOCORTICÓIDES INALATÓRIOS
Os GC inalatórios (fluticasona, budesonida, flunisolide e beclometasona) foram introduzidos no tratamento da asma há aproximadamente 30 anos e desde então ocupam uma função muito importante (82-84). Cerca de 80 a 90% da dose liberada pelos inaladores de GC são depositados na orofaringe e absorvidos posteriormente pelo trato gastro-intestinal (85). Após sua absorção, o GC faz sua 1ª passagem hepática, onde é convertido a formas inativas para excreção contribuindo pouco para os efeitos sistêmicos.
Cerca de 10 a 20% da dose liberada atinge os pulmões, onde exerce sua função terapêutica, e então é absorvida pela circulação, onde exerce maior efeito sistêmico.
Efeitos adversos dos glicocorticóides inalatórios
Os GC inalatórios podem apresentar efeitos colaterais localizados na orofaringe e laringe. Dentre eles: disfonia [relacionada à miopatia dos músculos laríngeos (86)], candidíase (87), tosse, hipertrofia de língua e aumento da sede (88).
No início da utilização dos GC inalatórios, acreditava-se que os pacientes estavam livres das complicações sistêmicas associadas à corticoterapia, mas, posteriormente, diversos casos de SC foram descritos na literatura em pacientes asmáticos que utilizavam GC inalatórios (89,90).
Os efeitos sistêmicos dos GC inalatórios variam conforme a dose administrada, a pneumopatia em tratamento e características individuais de cada paciente.
Assim como os GC sistêmicos, os GC inalatórios podem causar retardo de crescimento nas crianças, osteoporose, catarata e glaucoma (91-97).
Existem relatos apenas isolados de associação entre GC inalatórios e distúrbios psiquiátricos, dislipidemia, miopatia ou alterações da pele (98,99).
SÍNDROME DE RETIRADA DOS GLICOCORTICÓIDES
A descontinuação da terapia esteróide pode apresentar algumas dificuldades, dentre elas a possibilidade de supressão do eixo HHA resultando em insuficiência adrenal secundária, a possibilidades de piora da doença de base para a qual essa terapia foi iniciada e a chamada síndrome de retirada do esteróide (100). Esta última caracteriza-se por dificuldade além de muitos sintomas que o paciente pode enfrentar ao diminuir a dose ou suspender a terapia esteróide apesar de o eixo HHA demonstrar-se normal (101).
Supressão do eixo HHA
O tratamento com doses suprafisiológicas de GC, normalmente utilizado na prática clínica, suprime o eixo HHA, mas, na verdade, mesmo doses consideradas fisiológicas inibem a secreção de ACTH, sem, contudo, comprometer de forma acentuada a secreção de cortisol. Assim como a SC exógena, a dose e a duração da corticoterapia para que ocorra a supressão do eixo HHA varia entre os indivíduos.
Schaghicke e cols. (102) concluíram que nenhuma dose, duração ou cortisol plasmático basal podem predizer a função do eixo HHA. Mesmo nos pacientes que utilizaram doses equivalentes a 5 mg/d de prednisona tinham respostas suprimidas ou ausentes ao CRH.
Alguns autores acreditam que o uso de GC por menos de três semanas não levaria à supressão do eixo HHA (103). Já outros acreditam que doses supra-fisiológicas podem suprimir o eixo em apenas 5 dias. Sabe-se, no entanto, que o uso de doses fisiológicas por até 1 mês de duração não são capazes de suprimir o eixo HHA (104).
Muitas controvérsias também existem em relação ao tempo para a recuperação do eixo HHA, parecendo não haver correlação entre o tempo de corticoterapia e o período necessário para essa recuperação.
Na prática clínica há diversos testes disponíveis para se avaliar a função do eixo HHA.
Piora de doença da base
Pacientes portadores de doenças inflamatórias ou auto-imunes podem apresentar piora do quadro presente frente à diminuição ou retirada da terapia com GC.
Diante dessa situação, geralmente deve-se retornar à dose anterior à piora do quadro caso não existam contra-indicações (SC exógena grave, osteoporose com fraturas). Somente deve-se iniciar o desmame do GC quando o paciente apresentar-se estável de sua doença inicial e quando a terapia específica já estiver sendo administrada (tabela 3).
"Síndrome de retirada"
Os primeiros relatos de "síndrome de retirada" foram descritos por Amatruda em 1960 (105), tratando-se de patologia caracterizada por letargia, anorexia, náuseas, mialgia, cefaléia, perda de peso, febre, artralgia, descamação de pele, hipotensão postural e outros sinais e sintomas inespecíficos (100,101,105). É patologia pouco compreendida na prática clínica e sua atual prevalência não está determinada.
Apesar de tantos anos após sua descrição inicial, sua etiologia ainda não está totalmente esclarecida. Inicialmente, acreditava-se que a supressão do eixo HHA pelo hipercortisolismo endógeno ou exógeno era a principal responsável pelo quadro clínico, porém posteriormente observou-se que o eixo HHA encontra-se normal nesses pacientes (101). Subseqüentemente, Amatruda e cols. demonstraram algum grau de supressão do eixo HHA, porém os níveis de esteróides séricos e urinários eram normais, e que não haveria correlação entre o status do eixo HHA e a severidade e duração da síndrome de retirada (105). Dessa forma, essa condição passou a ser atribuída a um estado de "insuficiência adrenal relativa" dos tecidos que estavam expostos anteriormente a níveis elevados de GC por um longo período de tempo. Acredita-se que esses indivíduos desenvolveram certo grau de tolerância ao GC, tornando as doses fisiológicas geralmente utilizadas na reposição insuficientes para o sistema nervoso central e demais tecidos.
Independentemente da causa desses sintomas, esses pacientes não apresentam riscos cardiovasculares ou de insuficiência adrenal grave, e o manuseio da retirada do GC deve ser individualizado.
É importante ressaltar que muitos dos pacientes portadores dessa síndrome podem apresentar certo grau de dependência psicológica em relação ao GC.
A classificação em quatro subtipos para síndrome de retirada foi proposta por Dixon, em 1980, sendo caracterizada respectivamente como (106):
Tipo I. Evidências clínicas, bioquímicas e hormonais de supressão do eixo HHA, demonstradas de maneira rigorosa.
Tipo II. Recrudescência dos sintomas da doença de base para a qual a corticoterapia foi instalada, com eixo HHA normal.
Tipo III. Presença de sintomas característicos da síndrome, porém com resposta normal do eixo HHA e sem recrudescência da doença de base e cujos sintomas melhoram com o retorno da administração do GC. Esse subtipo geralmente apresenta síndrome de retirada verdadeira e pode estar associada a certa dependência física e/ou psicológica.
Tipo IV. Presença demonstrada de insuficiência do eixo HHA, porém sem sintomas da síndrome ou da doença de base.
Retirada do glicocorticóide
Apesar da ampla utilização da corticoterapia na prática clínica, não existem trials controlados a respeito do melhor esquema de retirada do GC.
Uma revisão recente (103) sobre os diversos esquemas de desmame da terapia com GC não encontrou suficientes evidências para se recomendar algum regime específico de retirada.
Qualquer plano de retirada deve-se basear em dois objetivos principais: 1) uso da menor dose possível capaz de controlar a doenças em questão e 2) uso da menor dose possível que não resulte em insuficiência adrenal secundária.
Geralmente, esquemas de retirada iniciam pela redução do GC a uma dose considerada fisiológica que é de aproximadamente 57,5 mg de prednisona ao dia, 1520 mg de hidrocortisona ao dia ou o equivalente. Durante essa fase, os pacientes não apresentam riscos de insuficiência adrenal, mas poderão evoluir com piora da doença de base (figura 3). O paciente já em uso de dose fisiológica pode realizar seu desmame de diversas maneiras e alguns autores preferem substituir o GC em uso para hidrocortisona pela vantagem de apresentar uma meia-vida menor, o que possibilita a recuperação mais rápida do eixo HHA. Outros recomendam utilizar o GC de ação intermediária como a prednisona em dias alternados e pela manhã, porém alguns pacientes não toleram o dia que não recebem a dose.
Durante esse estágio, é prudente realizar algumas medidas de cortisol sérico pela manhã como screening de insuficiência adrenal. Valores abaixo de 3,0 µg/dL indicam insuficiência e implicam em manutenção da terapia esteróide. Valores acima de 20,0 µg/dL indicam recuperação do eixo HHA (104) possibilitando a retirada do GC. Pacientes que apresentam níveis entre 3,0 e 20,0 µg/dL podem revelar uma produção de cortisol basal suficiente, mas não ideal para situações de estresse. Nesses casos, alguns testes para avaliação do eixo HHA devem ser realizados (figura 3).
De acordo com os resultados dos valores de cortisol nos testes de estímulo, a progressão do esquema deve ocorrer de maneira gradativa até que o paciente somente utilize GC nas situações de estresse durante um período de aproximadamente 1 ano após a retirada da terapia. Como já descrito anteriormente, o tempo necessário para a recuperação completa do eixo é extremamente variável e individual.
Situação especial
A chamada "síndrome de retirada" do GC foi inicialmente descrita para pacientes que pioram da doença de base ou que apresentam uma série de sintomas sugestivos de insuficiência adrenal quando a terapia esteróide é interrompida ou quando a dose utilizada é reduzida rapidamente. Uma forma menos reconhecida de síndrome de retirada do GC é aquela que se desenvolve em pacientes portadores de SC endógena após seu tratamento cirúrgico com sucesso apesar de níveis normais de cortisol plasmático. Nesses pacientes, apenas um pequeno aumento na dose de GC é capaz de melhorar o quadro clínico.
Papanicolaou e cols. demonstraram, em estudos recentes, que um aumento nos níveis de IL-6 está relacionado ao surgimento do quadro clínico da forma aguda da síndrome e que esse mesmo quadro pode ser obtido com a infusão endovenosa de IL-6 (107). Esses mesmos autores descreveram níveis de IL-6 ainda elevados no 9º- e 10º- dias pós-operatórios desses pacientes mesmo em reposição fisiológica de GC (108).
Recebido em 13/08/07
Aceito em 21/08/07
- 1. Shaker JL, Lukert BP. Osteoporosis associated with excess glucocorticoids. Endocrinol Metab Clin North Am 2005; 34(2):341-56.
- 2. Quddusi S, Browne P, Toivola B, Hirsch IB. Cushing syndrome due to surreptitious glucocorticoid administration. Arch Intern Med 1998;158-294.
- 3. Mann M, Koller E, Murgo A, Malozowski S, Bacsanyi J, Leinung M. Glucocorticoidlike activity of megestrol. A summary of Food and Drug Administration experience and a review of the literature. Arch Intern Med 1997; 157(15):1651-6.
- 4. Esteban S. Daily Cortisol Production rate in man determined by stable isotope dilution, mass spectrometry. J Clin Endocrinol Metab 1991;72:39-45.
- 5. Pensabeni-Jasper T, Panush RS. Review: corticosteroid usage: observations at a community hospital. Am J Med Sci 1996;234-311.
- 6. Yokoyama H, Takabatake T, Takaeda M, Wada T, Naito T, Ikeda K, et al. Up-regulated MHC-class II expression and gamma-IFN and soluble IL-2R in lupus nephritis. Kidney Int 1992;42:755.
- 7. Buttgereit F, Wehling M, Burmester GR. A new hypothesis of modular glucocorticoid actions. Arthritis Rheum 1998; 41:761.
- 8. Kehlet H, Binder C, Blichert-Toft M. Glucocorticoid maintenance therapy following adrenalectomy: assessment of dosage and preparation. Clin Endocrinol 1976;5:37.
- 9. Pensabeni-Jasper T, Panush RS. Review: corticosteroid usage: observations at a community hospital. Am J Med Sci 1996;234-311.
- 10. Walsh P, Aeling JL, Huff L, Weston WL. Hypothalamus-pituitary-adrenal axis suppression by superpotent topical steroids. J Am Acad Dermatol 1993;29:501.
- 11. Keipert JA, Kelly R. Temporary Cushing's syndrome from percutaneous absorption of betamethasone 17-valerate. Med J Austral 1971;1:542.
- 12. Feiwel M, James V, Barnett ES. Effect of potent topical steroids on plasma cortisol levels of infants and children with eczema. Lancet 1969;1:485.
- 13. Garber EK, Fan PT, Bluestone R. Realistic guidelines in corticosteroid therapy of rheumatic disease. Semin Arthritis Rheum 1981;11:231.
- 14. Laan RF, van Riel PL, van de Putte LB, van Erning LJ, van't Hof MA, Lemmens JA. Low-dose prednisone induces rapid reversible axial bone loss in patients with rheumatoid arthritis. Ann Intern Med 1993;119:963-8.
- 15. Allen DB, Julius JR, Breen TJ, Attie KM. Treatment of glucocorticoid-induced growth suppression with growth hormone. J Clin Endocrinol Metab 1998;83:2824.
- 16. Harter JG, Reddy WJ, Thorn GW. Studies on an intermittent corticosteroid dosage regimen. N Engl J Med 1963;269-591.
- 17. Faiçal SE, Uehara HM. Efeitos sistêmicos e síndrome de retirada em tomadores crônicos de corticosteróides. Rev Ass Med Brasil 1998;44(1):69-74.
- 18. Danowski TS, Bonessi JV, Sabeh G, Sutton RD, Webster MW Jr, Sarver ME. Probabilities of pituitary-adrenal responsiveness after steroid therapy. Ann Intern Med 1964;61:11-26.
- 19. Kaplan JG, Barasch E, Hirschfeld A, Ross L, Einberg K, Gordon M. Spinal epidural lipomatosis: a serious complication of iatrogenic Cushing's syndrome. Neurology 1989; 39(8):1031-4.
- 20. Kumar S, Singh RJ, Reed AM, Lteif AN. Cushing's syndrome after intra-articular and intradermal administration of triamcinolone acetonide in three pediatric patients. Pediatrics 2004;113(6):1820-4.
- 21. Teelucksingh S, Balkaran B, Ganeshmoorthi A, Arthur P. Prolonged childhood Cushing's syndrome secondary to intralesional triamcinolone acetonide. Ann Trop Paediatr 2002; 22(1):89-91.
- 22. Lavin PJ, Workman R. Cushing syndrome induced by serial occipital nerve blocks containing corticosteroids. Headache 2001;41(9):902-4.
- 23. Barlow AD, Clarke GA, Kelly MJ. Acute adrenal crisis in a patient treated with rectal steroids. Colorectal Dis 2004; 6(1):62-4.
- 24. De Wachter E, Vanbesien J, De Schutter I, Malfroot A, De Schepper J. Rapidly developing Cushing syndrome in a 4-year-old patient during combined treatment with itraconazole and inhaled budesonide. Eur J Pediatr 2003; 162(7-8):488-9.
- 25. Main KM, Skov M, Sillesen IB, Dige-Petersen H, Müller J, Koch C, et al. Cushing's syndrome due to pharmacological interaction in a cystic fibrosis patient. Acta Paediatr 2002; 91(9):1008-11.
- 26. Clevenbergh P, Corcostegui M, Gérard D, Hieronimus S, Mondain V, Chichmanian RM, et al. J Iatrogenic Cushing's syndrome in an HIV-infected patient treated with inhaled corticosteroids (fluticasone propionate) and low dose ritonavir enhanced PI containing regimen. Infect 2002; 44(3):194-5.
- 27. Gupta SK, Dubé MP. Exogenous Cushing syndrome mimicking human immunodeficiency virus lipodystrophy. Clin Infect Dis 2002;15;35(6):69-71.
- 28. Fries JF, Williams CA, Ramey DR, Bloch DA. The relative toxicity of alternative therapies for rheumatoid arthritis: implications for the therapeutic progression. Semin Arthritis Rheum 1993;23:68-73.
- 29. Karagas MR, Cushing GL Jr, Greenberg ER, Mott LA, Spencer SK, Nierenberg DW. Non-melanoma skin cancers and glucocorticoid therapy. Br J Cancer 2001;85(5):683-6.
- 30. Sorensen HT, Mellemkjaer L, Nielsen GL, Baron JA, Olsen JH, Karagas MR. Skin cancers and non-Hodgkin lymphoma among users of systemic glucocorticoids: a population-based cohort study. J Natl Cancer Inst 2004; 96(9):709-11.
- 31. Tyrrel JB, Baxter JD. Glucocorticoid therapy. In: Felig P, Baxter JD, Broadus AE et al. Endocrinology and Metabology New York: McGraw-Hill Book, 1988 pp. 788-817.
- 32. Polansky JR, Weinreb RM. Anti-inflammatory agents: steroids as anti-inflammatory agents. In: Sears ML. Handbook of experimental pharmacology vol. 69. New York: Springer-Verlag, 1984 p. 459.
- 33. Manabe S, Bucala R, Cerami A. Nonenzymatic addition of glucocorticoids to lens proteins in steroid-induced cataracts. J Clin Invest 1984;74:1803.
- 34. Berkowitz JS, David DS, Sakai S, Shoji H, Cheigh JS, Riggio RR, et al. Ocular complications in renal transplant recipients. Am J Med 1973;55(3):492-5.
- 35. Skalka HW, Prchal JT. Effect of corticosteroids on cataract formation. Arch Ophthalmol 1980;98(10):1773-7.
- 36. Baxter JD. Glucocorticoid hormone action. In: Gill GN. Pharmacology of adrenal cortical hormones Oxford: Pergamon, 1979 p. 67.
- 37. Exton JH. Regulation of gluconeogenesis by glucocorticoids. In: Baxter JD, Rousseau GG. Glucocorticoid hormone action New York: Springer-Verlag, 1979 p. 535.
- 38. Long WF. A case of elevated intraocular pressure associated with systemic steroid therapy. Am J Optom Physiol Opt 1977;54(4):248-52.
- 39. Karadimas P, Kapetanios A, Bouzas EA. Central serous chorioretinopathy after local application of glucocorticoids for skin disorders. Arch Ophthalmol 2004;122(5):784-6.
- 40. Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med 2004;141(10):764-70.
- 41. Souverein PC, Berard A, Van Staa TP, Cooper C, Egberts AC, Leufkens HG, et al. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart 2004;90(8):859-65.
- 42. Taskinen MR, Nikkila EA, Pelkonen R, Sane T. Plasma lipoproteins, lipolytic enzymes, and very low density lipoprotein triglyceride turnover in Cushing's syndrome. J Clin Endocrinol Metab 1983;57:619.
- 43. Berg AL, Nilsson-Ehle P. ACTH lowers serum lipids in steroid-treated hyperlipemic patients with kidney disease. Kidney Int 1996;50(2):538-42.
- 44. Bollet AJ, Black R, Bunim JJ. Major undesirable side-effects resulting from prednisolone and prednisone. J Am Med Assoc 1955;158(6):459-63.
- 45. Whitworth JA. Adrenocorticotrophin and steroid-induced hypertension in humans. Kidney Int 1992;37(suppl):34-7.
- 46. Saruta T, Suzuki H, Handa M, Igarashi Y, Kondo K, Senba S. Multiple factors contribute to the pathogenesis of hypertension in Cushing's syndrome. Clin Endocrinol Metab 1986;62(2):275-9.
- 47. Jackson SH, Beevers DG, Myers K. Does long-term low-dose corticosteroid therapy cause hypertension? Clin Sci (Lond) 1981;7:381-3.
- 48. Hsueh AJ, Erickson GF. Glucocorticoid inhibition of FSH induced estrogen production in cultured rat granulosa cells. Steroids 1978;32:639-48.
- 49. Gebhard KL, Maibach HI. Relationship between systemic corticosteroids and osteonecrosis. Am J Clin Dermato 2001;2(6):377-88.
- 50. Felson DT, Anderson JJ. Across-study evaluation of association between steroid dose and bolus steroids and avascular necrosis of bone. Lancet 1987;1(8538):902-6.
- 51. Mankin HJ. Nontraumatic necrosis of bone (osteonecrosis). N Engl J Med 1992;326(22):1473-9.
- 52. Baxter JD. Mechanisms of glucocorticoid inhibition of growth. Kidney Int 1978;14-330.
- 53. Loeb JN. Corticosteroids and growth. N Engl J Med 1976; 295-547.
- 54. Dempster DW, Arlot MA, Meunier PJ. Mean wall thickness and formation periods of trabecular bone packets in corticosteroid-induced osteroporosis. Calcif Tissue Int 1983; 35:410-7.
- 55. Reid IR, Katz JM, Ibbertson HK, Gray DH. The effects of hydrocortisone, parathyroid hormone, and the bisphosphonate, APD, on bone resorption in neonatal mouse calvaria. Calcif Tissue Int 1986;38:38-43.
- 56. Gronowicz G, McCarthy MB, Woodiel F, Raisz LG. Effects of corticosterone and parathyroid hormone on formation and resorption in cultured fetal rat parietal bones. Am Soc Bone Min Res 1988;3:5-114.
- 57. Lukert BP, Raisz LR. Glucorticoid-induced osteoporosis: pathogenesis and management. Ann Intern Med 1990; 112:352-64.
- 58. Hahn TJ, Halstead LR, Strates B, Imbimbo B, Baram DT. Comparison of subacute effects of oxazacort and prednisolone on mineral metabolism in man. Calcif Tissue Int 1980;30:109-15.
- 59. Delorme AC, Danan JL, Mathieu H. Biochemical evidence for the presence of two vitamin D dependent calcium-binding proteins in mouse kidney. Biochem Chem 1993;258:1878-84.
- 60. Meunier PJ, Bressot C. Endocrine influences on bone cells and bone remodeling evaluated by clinical histomorphometry. In: Parsons JA (ed). Endocrinology of calcium metabolism New York: Raven Press, 1982 pp. 445-65.
- 61. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res 2000;15(6):993-1000.
- 62. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology 2000;39(12):1383-9.
- 63. Laan RF, Buijs WC, van Erning LJ, Lemmens JA, Corstens FH, Ruijs SH, et al. Differential effects of glucocorticoids on cortical appendicular and cortical vertebral bone mineral content. Calcif Tissue Int 1993;52(1):5-9.
- 64. Kumagai S, Kawano S, Atsumi T, Inokuma S, Okada Y, Kanai Y, et al. J. Vertebral fracture and bone mineral density in women receiving high dose glucocorticoids for treatment of autoimmune diseases. Rheumatol 2005;32(7):1414.
- 65. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest 1998;102(2):274-82.
- 66. Fain JN. Inhibition of glucose transport in fat cells and activation of lipolysis by glucocorticoids. In: Baxter JD, Rousseau GG. Glucocorticoid hormone action New York: Springer-Verlag, 1979 p. 7.
- 67. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days' corticosteroid treatment. A prospective study. Psychoneuroendocrinology 1996; 21(1):25-31.
- 68. Minden SL, Orav J, Schildkraut JJ. Hypomanic reactions to ACTH and prednisone treatment for multiple sclerosis. Neurology 1988;38(10):1631-4.
- 69. Goggans FC, Weisberg LJ, Koran LM. Lithium prophylaxis of prednisone psychosis: a case report. J Clin Psychiatry 1983;44(3):111-2.
- 70. Kershner P, Wang-Cheng R. Psychiatric side effects of steroid therapy. Psychosomatics 1989;30(2):135-9.
- 71. Chau SY, Mok CC. Factors predictive of corticosteroid psychosis in patients with systemic lupus erythematosus. Neurology 2003;61(1):104-7.
- 72. Gilin JC, Jacobs LS, Fram DH, Snyder F. Acute effect of a glucocorticoid on normal human sleep. Nature 1972; 237-398.
- 73. Balasubramanian S, Shivbalan S, Ganesh R. Pseudotumor cerebri following oral betamethasone for common cold. Indian J Pediatr 2005;72(11):994.
- 74. Lenzen S, Bailey CJ. Thyroid hormones, gonadal and adrenocortical steroids and the function of the islets of Langerhans. Endocrinol Rev 1984;5:411.
- 75. Olefsky JM, Kimmerling G. Effects of glucocorticoids on carbohydrate metabolism. Am J Med Sci 1976;271(2):202-10.
- 76. Miller SE, Neilson JM. Clinical features of the diabetic syndrome appearing after ateroid therapy. Postgrad Med J 1964;40:660-9.
- 77. Fahey JV, Guyre PM, Munck A. Mechanisms of anti-inflammatory actions of glucocorticoids. In: Weissman G. Advances in inflammatory research, vol 2. New York: Raven, 1981 p. 21.
- 78. Meduri GU, Golden E, Freire AX, Taylor E, Zaman M, Carson SJ, et al. Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest 2007; 131(4):954-63.
- 79. Sendi P, Wolf A, Graber P, Zimmerli W. Multiple opportunistic infections after high-dose steroid therapy for giant cell arteritis in a patient previously treated with a purine analog. Scand J Infect Dis 2006;38(10):922-4.
- 80. Ly HT, Thermidor M, Cunha BA. Pneumocystis carinii pneumonia in a non-HIV patient on steroids. Heart Lung 2004;33(4):261-4.
- 81. Sampson PA, Brooke BN, Winstone NE. Biochemical confirmation of collapse due to adrenal failure. Lancet 1961; 24:1-1377.
- 82. Barnes PJ, Pedersen S, Busse WW. Efficacy and safety of inhaled corticosteroids. New developments. Am J Respir Crit Care Med 1998;157:S1.
- 83. Geddes DM. Inhaled corticosteroids: Benefits and risks. Thorax 1992;47:404.
- 84. Toogood JH. Complications of topical steroid therapy for asthma. Am Rev Respir Dis 1990;14(part 2):S89.
- 85. Johnson M. Pharmacodynamics and pharmacokinetics of inhaled glucocorticoids. J Allergy Clin Immunol 1996; 97:169.
- 86. Williamson IJ, Matusiewicz SP, Brown PH, Greening AP, Crompton GK. Frequency of voice problems and cough in patients using pressurized aerosol inhaled steroid preparations. Eur Respir J 1995;8:590-4.
- 87. Toogood JH, Jennings B, Greenway RW, Chuang L. Candidiasis and dysphonia complicating beclomethasone treatment of asthma. J Allergy Clin Immunol 1980; 65:145.
- 88. Roland NJ, Bhalla RK, Earis J. The local side effects of inhaled corticosteroids: current understanding and review of the literature. Chest 2004;126:213.
- 89. Curtis JR, Westfall AO, Allison JJ, Freeman A, Saag KG. Channeling and adherence with alendronate and risedronate among chronic glucocorticoid users. Osteoporos Int 2006; 17(8):1268-74.
- 90. Pouw EM, Prummel MF, Oosting H, Roos CM, Endert E. Beclomethasone inhalation decreases serum osteocalcin concentrations. BMJ 1991;302(6777):627-8.
- 91. Thomas BC, Stanhope R, Grant DB. Impaired growth in children with asthma during treatment with conventional doses of inhaled corticosteroids. Acta Paediatr 1994;83:196.
- 92. Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000;343:1064.
- 93. Israel E, Banerjee TR, Fitzmaurice GM, Kotlov TV, LaHive K, LeBoff MS. Effects of inhaled glucocorticoids on bone density in premenopausal women. N Engl J Med 2001; 345:941-7.
- 94. Hubbard R, Tattersfield A, Smith C, West J, Smeeth L, Fletcher A. Use of inhaled corticosteroids and the risk of fracture. Chest 2006;130:1082-8.
- 95. White M, Crisalida T, Li H, Economides A, Kaliner M. Effects of long-term inhaled corticosteroids on adrenal function in patients with asthma. Ann Allergy Asthma Immunol 2006;96:437-44.
- 96. Mitchell P, Cumming RG, Mackey DA. Inhaled corticosteroids, family history, and risk of glaucoma. Ophthalmology 1999; 106:2301.
- 97. Garbe E, Suissa S, LeLorier J. Association of inhaled corticosteroid use with cataract extraction in elderly patients. JAMA 1998;280:539.
- 98. Roy A, Leblanc C, Paquette L, Ghezzo H, Côté J, Cartíer A, et al. Skin bruising in asthmatic subjects treated with high doses of inhaled steroids: frequency and association with adrenal function. Eur Respir J 1996;9:226-31.
- 99. Herzog AG. Proximal myopathy associated with inhaled steroids. JAMA 1999;281:37.
- 100.Hochberg Z, Pacak K, Chrousos GP. Endocrine withdrawal syndromes. Endocr Rev 2003;24(4):523-38.
- 101.Amatruda TT, Hollingsworth DR, D'Esopo ND, Upton GV, Bondy PK. A study of the mechanism of the steroid withdrawal syndrome. Evidence for integrity of the hypothalamic-pituitary-adrenal system. J Clin Endocrinol Metab 1960; 20:339-54.
- 102.Schlaghecke R, Kornely E, Santen RT, Ridderskamp P. The effect of long-term glucocorticoid therapy on pituitary-adrenal responses to exogenous corticotropin-releasing hormone. N Engl J Med 1992;326(4):226-30.
- 103.Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. Endocrinol Metab Clin North Am 2002;31(3):751-78.
- 104.Krasner AS. Glucocorticoid-induced adrenal insufficiency. JAMA 1999;282(7):671-6.
- 105.Amatruda Jr TT, Hurst MM, D'Esopo ND. Certain endocrine and metabolic facets of the steroid withdrawal syndrome. J Clin Endocrinol Metab 1965;25:1-207.
- 106.Dixon RB, Christy NP. On the various forms of corticosteroid withdrawal syndrome. Am J Med 1980;68:224.
- 107.Papanicolaou DA, Tsigos C, Oldfield EH, Chrousos GP. Acute glucocorticoid deficiency is associated with plasma elevations of interleukin-6: does the latter participate in the symptomatology of the steroid withdrawal syndrome and adrenal insufficiency? J Clin Endocrinol Metab 1996; 81(6):2303-6.
- 108.Papanicolaou DA, Wilder RL, Manolagas SC, Chrousos GP. The pathophysiologic roles of interleukin-6 in human disease. Ann Intern Med 1998;128(2):127-37.
Endereço para correspondência:
Datas de Publicação
-
Publicação nesta coleção
14 Jan 2008 -
Data do Fascículo
Nov 2007
Histórico
-
Aceito
21 Ago 2007 -
Recebido
13 Ago 2007