Abstracts
Mutations in the glucokinase gene (GCK) account for many cases of monogenic diabetes featuring maturity-onset diabetes of the young type 2 (MODY2). The clinical pattern of this form of hyperglycemia is rather stable, with a slight elevation in blood glucose, which is usually not progressive. Patients rarely require pharmacological interventions and microvascular complications related to diabetes are unusual. We describe the clinical follow-up of two cases of MODY2 with two different mutations in GCK gene, one in exon 7, p.Glu265Lys (c.793 G> A), which has been previously described, and a novel one, in exon 2, p.Ser69Stop (c. 206C> G). The clinical course of both cases shows similarity in metabolic control of this form of diabetes over the years. Arq Bras Endocrinol Metab. 2012;56(8):490-5
Mutações no gene da glicoquinase (GCK) são determinantes de uma forma de diabetes monogênico denominada de MODY2 (maturity-onset diabetes of the young, tipo 2). O padrão clínico dessa forma de distúrbio glicêmico é bastante estável, com hiperglicemia leve, geralmente não progressiva. Intervenções farmacológicas raramente são necessárias e complicações crônicas secundárias ao diabetes são infrequentes. Descrevemos o acompanhamento clínico de dois casos de MODY2 com duas mutações diferentes, uma no éxon 7, p.Glu265Lys (c.793 G>A) já descrita anteriormente, e outra inédita no éxon 2 p.Ser69Stop (c. 206C>G). A evolução clínica de ambos os casos demonstra uma semelhança no padrão metabólico dessa forma de diabetes ao longo dos anos. Arq Bras Endocrinol Metab. 2012;56(8):490-5
CASE REPORT
Clinical follow-up of two Brazilian subjects with glucokinase-MODY (MODY2) with description of a novel mutation
Seguimento clínico de dois pacientes brasileiros com MODY-glicoquinase (MODY2) e descrição de uma nova mutação
Thais DellaMannaI; Magnus R. da SilvaII; Antonio Roberto ChacraIII; Ilda S. KuniiII; Ana Luiza RolimII; Gilberto FuruzawaII; Rui Monteiro de Barros MacielII; André Fernandes ReisII,III
IInstituto da Criança, Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo (IPq-HC-FMUSP), São Paulo, SP, Brazil
IILaboratório de Endocrinologia Molecular e Translacional, Escola Paulista de Medicina, Universidade Federal de São Paulo (Unifesp-EPM), São Paulo, SP, Brazil
IIICentro de Diabetes, Unifesp-EPM, São Paulo, SP, Brazil
Correspondence Correspondence to: André Fernandes Reis Escola Paulista de Medicina, Universidade Federal de São Paulo Rua Pedro de Toledo, 669, 11º andar 04039-032 São Paulo, SP, Brazil andrefreis@terra.com.br
SUMMARY
Mutations in the glucokinase gene (GCK) account for many cases of monogenic diabetes featuring maturity-onset diabetes of the young type 2 (MODY2). The clinical pattern of this form of hyperglycemia is rather stable, with a slight elevation in blood glucose, which is usually not progressive. Patients rarely require pharmacological interventions and microvascular complications related to diabetes are unusual. We describe the clinical follow-up of two cases of MODY2 with two different mutations in GCK gene, one in exon 7, p.Glu265Lys (c.793 G> A), which has been previously described, and a novel one, in exon 2, p.Ser69Stop (c. 206C> G). The clinical course of both cases shows similarity in metabolic control of this form of diabetes over the years. Arq Bras Endocrinol Metab. 2012;56(8):490-5
SUMÁRIO
Mutações no gene da glicoquinase (GCK) são determinantes de uma forma de diabetes monogênico denominada de MODY2 (maturity-onset diabetes of the young, tipo 2). O padrão clínico dessa forma de distúrbio glicêmico é bastante estável, com hiperglicemia leve, geralmente não progressiva. Intervenções farmacológicas raramente são necessárias e complicações crônicas secundárias ao diabetes são infrequentes. Descrevemos o acompanhamento clínico de dois casos de MODY2 com duas mutações diferentes, uma no éxon 7, p.Glu265Lys (c.793 G>A) já descrita anteriormente, e outra inédita no éxon 2 p.Ser69Stop (c. 206C>G). A evolução clínica de ambos os casos demonstra uma semelhança no padrão metabólico dessa forma de diabetes ao longo dos anos. Arq Bras Endocrinol Metab. 2012;56(8):490-5
INTRODUCTION
Maturity onset diabetes of the young (MODY) refers to a group of clinically heterogeneous hereditary forms of diabetes that are defined at the molecular genetic level by mutations in different genes. All subtypes show beta-cell dysfunction associated with defects in insulin secretion, usually with young-onset of diabetes (childhood, adolescence, or young adults), autosomal dominant inheritance (presence of three generations of the same affected lineage) with variable clinical features depending on the mutated gene (1-3). MODY is one of the monogenic forms of diabetes to be distinguished from the polygenic types of diabetes, especially type 1 and type 2, which involve more complex combinations of genetic and environmental factors. At least seven forms of MODY have been described by molecular genetic studies of diabetes, and are related to mutations in Glucokinase (GCK), HNF1A, HNF4A, HNF1B, NEUROD1, PDX1, and INS genes, with different clinical presentations. Identification of a MODY mutation is important for correct diagnosis, appropriate genetic counseling, evaluation of prognosis, and selection of the best treatment (4,5). MODY is rare, with an estimated prevalence between 0.3% and 2.4% of the diabetes cases, although these figures are considered underestimations; mutations in HNF1A and GCK genes account for approximately 70% of cases in all populations studied (3,6), including in Brazil (7-9).
Glucokinase (GCK) belongs to the hexokinase family of enzymes, and catalyses the conversion of glucose to glucose-6-phosphate in the first reaction of the glycolytic pathway (10). GCK plays a major role in the regulation of insulin release and has been termed the glucose sensor of pancreatic beta-cells because of its ability to alter glucose phosphorylation rates over a range of physiological glucose concentrations (11). The key regulatory role of GCK in the pancreatic beta-cells explains why mutations in this gene can result in both hyper- and hypoglycemia. For instance, gain-of-function heterozygous GCK mutations have been identified in patients with congenital hyperinsulinism (CHI) characterized by inappropriate oversecretion of insulin despite hypoglycemia (12). Meanwhile, a total of twelve GCK loss-of-function mutations have been reported in permanent neonatal diabetes mellitus (PNDM).
The clinical features that suggest the diagnosis of MODY2 are: a) persistent and stable fasting hyperglycemia over a period of months or years; b) glycated hemoglobin (HbA1c) frequently just below or just above the upper limit of normal; c) small blood glucose increment (less than 60 mg/dL) after an oral glucose overload test (2hr glucose fasting glucose); d) parents may have diabetes or at least one parent may have mildly raised fasting blood glucose; it is important to test fasting glucose in apparently unaffected parents (13,14). Existence of heterogeneity in the clinical phenotype of patients carrying GCK mutations has already been described, including uncommon presentations such as overt neonatal hyperglycemia, marked postprandial hyperglycemia, glucosuria, and even ketoacidosis (15-17). However, overall, the pattern of mild hyperglycemia is usually quite stable in most patients. Microvascular complications are very unusual in GCK-MODY, and pharmacological treatment is rarely needed, since most cases are managed by diet alone (5,13,18,19).
MODY diagnosis is defined by molecular genetics and predicts disease prognosis, besides identifying risk of diabetes in family members. However, genetic testing is expensive, so careful consideration is required when determining which patients should be tested (6).
The aim of this study was to report the clinical follow-up of two Brazilian patients with heterozygous GCK mutations, since the natural course of MODY2 is still unknown within our population. We also described a new mutation in GCK gene.
PATIENTS AND METHODS
Clinical and laboratory data were collected from each patient's records. The following biochemical parameters were measured on fasting blood samples: plasma glucose (FPG) by enzymatic method; cholesterol and triglycerides by colorimetric enzymatic method; HbA1c by high performance liquid chromatography (HPLC); serum fasting insulin, serum fasting C-peptide, anti-glutamic acid decarboxylase (GADA), anti-islet cell (ICA512) and anti-insulin (IAA) by radioimmunoassay. Oral glucose tolerance test (OGTT) was evaluated in fasting blood samples, and 2 hours after an oral glucose dose of 0.75 g/kg body weight (maximum 75 g). Homeostasis model assessment (HOMA) indexes of beta-cell function (%B) and insulin sensitivity (%S) were computed with the HOMA Calculator version 2.2 (20).
Genetic testing of case 1 was performed in Exeter, UK, and of case 2, in Unifesp, Brazil. Genomic DNA was extracted from peripheral blood leukocytes by standard procedures; exons 1A-10 and exon-intron boundaries of the GCK gene were screened for mutation by direct sequencing using Big Dye Terminator on ABI PRISM 3131XL Genetic Analyzer (Applied Biosystems, UK). Polymerase chain reaction (PCR) primers were supplied on demand.
Parents of both subjects gave their written informed consent for genetic analysis and disclosure of the results. The research was approved by the Ethics Committee of Escola Paulista de Medicina, Unifesp.
Patient 1
The index patient was a boy, the only child to non-consanguineous parents, born at term with a birth weight of 2.900 g (SDS -1.08) and length of 48 cm (SDS -0.74), whose growth and neurological development were normal. At 11 years of age, during laboratory tests for the investigation of tonsillitis, he showed fasting blood glucose level of 125 mg/dL. The test was repeated a few days later and resulted in 123 mg/dL. OGTT was performed with the following values: fasting, 112 mg/dL and 2 h post-load: 151 mg/dL, consistent with the American Diabetes Association (ADA) criteria for impaired fasting glucose and impaired glucose tolerance (pre-diabetes) (21). His pubertal development was normal for age, and no other health problems were disclosed in physical examination. In his family history, the father reported impaired fasting blood glucose level of 110 mg/dL, which could not be confirmed, and cases of diabetes were denied.
Laboratory investigation demonstrated negative results for anti-glutamic acid decarboxylase (GADA), anti-islet cell (ICA512) and anti-insulin (IAA) self-antibodies. Fasting C-peptide levels showed good pancreatic reserve (1.4 ng/mL; reference value: 1.1-4.4 ng/mL), and HbA1C was 6.1% (reference value: 4.8-5.9%). Based on his clinical presentation, the hypothesis of MODY2 was formed. Sequencing of the GCK gene identified a mutation in exon 7, p.Glu265Lys (c.793 G>A). The patient was advised to perform regular physical activity for weight maintenance. Clinical follow-up demonstrated stable fasting blood glucose, and HbA1C levels around 6.3% (ranging from 5.9% to 6.8%; HPLC) in a period of more than 9 years. He had normal pubertal development with no worsening of glycemic control (Table 1). No medication was prescribed throughout this period. The patient remained very active, always practicing regular physical activity, maintaining normal weight and body mass index (BMI). His parents did not want to perform genetic tests.
Patient 2
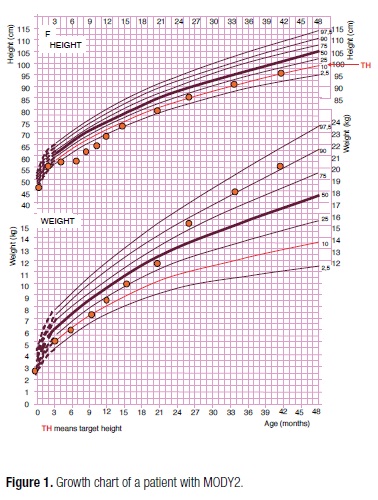
The proband was a girl, the second child of a non-consanguineous parents, born at 38 weeks of gestation, with a birth weight of 2.740 g (SDS -1.29), length of 46 cm (SDS -1.44). The mother was diagnosed with hypothyroidism and gestational diabetes, and treated with levothyroxine and insulin; she remained diabetic after delivery, with irregular use of metformin. Her father was obese and the maternal great-grandmother, grandmother, and grandfather had diabetes treated with sulphonylurea. The postnatal course was uncomplicated but growth retardation was observed during the first 6 months of life, and mild hyperglycemia was detected in the laboratory work-up (Table 2). At 18 months of age, type 1 diabetes-associated self-antibodies (ICA512, GADA, IAA) were negative, C-peptide level was 0.7 ng/mL, HbA1c was 6.6% and fasting blood glucose was 136 mg/dL. The hypothesis of MODY2 was formed, and the family was advised about the importance of a healthy lifestyle with balanced diet and regular physical activity for better prognosis. However, after 2 years of age, she was started on cafeteria food and her weight gain was progressively greater, despite her growth rate remaining stable, in the familial growth pattern (SDS -1.74) (Figure 1). Some episodes of postprandial hyperglycemia (197; 180; 201 mg/dL) were detected by self-monitoring of blood glucose, without glucosuria. Serial OGTTs were performed and the results were consistent with the ADA criteria for impaired fasting glucose and impaired glucose tolerance (pre-diabetes) in most occasions (21) (Table 2). During her clinical follow-up throughout the next 8 years, she maintained fasting blood glucose between 117-132 mg/dL and HbA1c between 6.6%-6.8% (Table 2). Recently, she started pubertal development (Tanner B1P2) at the age of 10 years and 3 months, with bone age of 11 years. Sequencing of the GCK has recently identified a nonsense mutation p.Ser69Stop (c. 206C>G) in the proband and her mother. Her father, without history of diabetes, showed no mutation.
DISCUSSION
We describe here the clinical follow-up of two GCK-MODY pediatric patients whose metabolic control was uneventful without diabetes-related complications. Molecular analysis demonstrated a missense mutation p.Glu265Lys (c.793 G>A) in patient 1, which has already been described in Italian, Spanish, and British populations (22-24). Previous functional studies demonstrated that this mutation strongly affects protein stability (25). In patient 2, a novel nonsense mutation p.Ser69Stop (c. 206C>G) in exon 2 was identified in the patient and her mother, thus demonstrating the co-segregation of this mutation and diabetes mellitus phenotype.
A total of 620 GCK mutations have been identified in 1,441 families with PNDM, MODY, or CHI. Mutations are distributed throughout the gene and no common mutations or "hot spots" have been found (24). All MODY2 cases are made up of heterozygous for nonsense, missense, or frameshift mutations and result in a deficiency of GCK activity (26-29). More than 50 mutations were functionally characterized, and most of them alter the kinetics of the enzyme. In other mutations where kinetics was normal, the mutational mechanism may be related to the effect on enzyme stability (as in our case 1), or binding to regulatory molecules (24). There is no clear association between phenotype (i.e., insulin secretion) and genotype (including type, topography) in GCK-MODY patients (30).
In an 11-year period of follow-up, it was observed that glucose tolerance of MODY2 adult subjects may remain stable for many years, probably because of the stability of the glucokinase-related beta-cell defect. However, if or when insulin resistance develops, the beta-cell defect may impair the increase in insulin secretion, and may result in deterioration of glucose tolerance (31). Of note, our patient 2 became obese and there was a noticeable variation in insulin sensitivity measured by HOMAIR/HOMAS. However, the index of insulin secretion HOMAbeta was persistently less than 100%, even when HOMAIR/HOMAS was 2.3/42.6%, suggesting failure in insulin secretion (Table 2). In both cases, but particularly in case 1, the stability of C-peptide plasma levels all over this period suggests stability of pancreatic beta-cell reserve in MODY2 (Table 1), in contrast with the findings commonly observed in patients with DM2, and especially with DM1 (32).
It has been recently demonstrated in a cohort of healthy children aged 5-14 years who were enrolled in the Early Bird Diabetes study, that insulin resistance starts to rise in mid-childhood, some years before puberty, mostly independent of rising adiposity and insulin-simile growth factor-1 (IGF-1) (33). The condition of Patient 1 remained under control throughout his pubertal development, reinforcing the concept that hyperglycemia in MODY2 remains stable over time. Patient 2 has only recently initiated pubarche.
Another interesting aspect is the existence of a possible relationship between GCK mutation and growth retardation. Failure to thrive motivated the reference of Patient 2 to further investigation. Intrauterine growth retardation and reduced birth weight are clinical manifestations frequently described in infants with permanent neonatal diabetes mellitus due to mutations in KCNJ11, ABCC8 (34), INS (35,36), PDX1 (37) genes, and in homozygous GCK mutations in children within the first few days of life (26,29), as a reflection of insulin deficiency in utero. Studies with GCK-MODY pedigrees demonstrated that GCK-MODY newborns are lighter than their unaffected siblings, independent of the parental origin of the mutation (38-40). Hattersley and cols. (41) analyzed the impact caused by a heterozygous mutation in the GCK gene in fetal growth and birth weight of 58 children from 10 extended GCK pedigrees, and observed that the inheritance of a GCK mutation by the fetus resulted in mean birth weight reduction of 533 g (p = 0.002). Length at birth was also reported in 26 children, and the presence of a fetal GCK mutation significantly reduced length (50.3 ± 5.3 vs. 55.5 ± 2.3 cm; p = 0.009). These studies brought evidence that birth weight is also determined by fetal genotype. Velho and cols. demonstrated that a large proportion of babies show early postnatal catch-up after birth, as observed in our case 2. Also, they found that neither maternal nor fetal mutations alter adult height or weight of the offspring (42).
In conclusion, we described two Brazilian patients carrying mutations in the GCK gene, whose 9-year clinical follow-up confirmed the stability of metabolic control and absence of vascular complications without antidiabetic medications. We also report a novel nonsense mutation p.Ser69Stop (c. 206C>G) associated with the MODY2 phenotype. This case report reinforces the importance of genetic diagnosis of monogenic forms of diabetes in order to establish prognosis and appropriate treatment intervention.
Acknowledgments: the authors are grateful to Sian Ellard, from Peninsula Medical School Molecular Genetics Laboratory-UK, who performed genetic testing of case 1.
Disclosure: no potential conflict of interest relevant to this article was reported.
Received on June/21/2012
Accepted on Sept/10/2012
- 1. Velho G, Froguel P. Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young. Eur J Endocrinol. 1998;138:233-9.
- 2. Oliveira CSV, Furuzawa GK, Reis AF. Diabetes mellitus tipo MODY. Arq Bras Endocrinol Metab. 2002;46(2):186-92.
- 3. Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504-8.
- 4. Njølstad PR, Molven A. To test, or not to test: time for a MODY calculator? Diabetologia. 2012;55:1231-4.
- 5. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:200-13.
- 6. Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia. 2012;55:1265-72.
- 7. Furuzawa GK, Giuffrida FM, Oliveira CS, Chacra AR, Dib SA, Reis AF. Low prevalence of MODY2 and MODY3 mutations in Brazilian individuals with clinical MODY phenotype. Diabetes Res Clin Pract. 2008;81(3):e12-4.
- 8. Moises RS, Reis AF, Morel V, Chacra AR, Dib SA, Bellanne-Chantelot C, Velho G. Prevalence of maturity-onset diabetes of the young mutations in Brazilian families with autosomal-dominant early-onset type 2 diabetes. Diabetes Care. 2001;24(4):786-8.
- 9. Maraschin J de F, Kannengiesser C, Murussi N, Campagnolo N, Canani LH, Gross JL, et al. HNF1alpha mutations are present in half of clinically defined MODY patients in South-Brazilian individuals. Arq Bras Endocrinol Metabol. 2008;52(8):1326-31.
- 10. Gloyn AL. Glucokinase (GCK) mutations in hyper and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat. 2003;22:353-36.
- 11. Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51(Suppl.3):S394-404.
- 12. Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226-30.
- 13. Stride A, Vaxillaire M, Tuomi T, Barbetti F, Njølstad PR, Hansen T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45:427-35.
- 14. Hattersley A, Bruining J, Shiel J, Njolstad P, Donaghue KC. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatric Diabetes. 2009;10(Suppl.12):33-42.
- 15. Fajans SS, Bell GI. Phenotypic heterogeneity between different mutations of MODY subtypes and within MODY pedigrees. Diabetologia. 2006;49:1106-8.
- 16. Cuesta-Muñoz AL, Tuomi T, Cobo-Vuilleumier N, Koskela H, Odili S, Stride A, et al. Clinical heterogeneity in monogenic diabetes caused by mutations in the glucokinase gene (GCK-MODY). Diabetes Care. 2010;33(2):290-2.
- 17. Borowiec M, Mysliwiec M, Fendler W, Antosik K, Brandt A, Malecki M, et al. Phenotype variability and neonatal diabetes in a large family with heterozygous mutation of the glucokinase gene. Acta Diabetol. 2011;48:203-8.
- 18. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971-80.
- 19. Ellard S, Bellanné-Chantelot C, Hattersley AT; the European Molecular Genetics Quality Network (EMQN) MODY group. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia. 2008;51:546-53.
-
20HOMA Calculator. HOMA2 model. Available at: http://www.dtu.ox.ac.uk/index.php?maindoc/homa/index.php Accessed on: May 1, 2012.
- 21. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004;27:S5-10.
- 22. Estalella I, Rica I, Perez de Nanclares G, Bilbao JR, Vazquez JA, San Pedro JI, et al.; Spanish MODY Group. Mutations in GCK and HNF-1alpha explain the majority of cases with clinical diagnosis of MODY in Spain. Clin Endocrinol (Oxf). 2007;67(4):538-46.
- 23. Tinto N, Zagari A, Capuano M, De Simone A, Capobianco V, Daniele G, et al. Glucokinase gene mutations: structural and genotype-phenotype analyses in MODY children from South Italy. PLoS One. 2008;3(4):e1870.
- 24. Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512-26.
- 25. Galán M, Vincent O, Roncero I, Azriel S, Boix-Pallares P, Delgado-Alvarez E, et al. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem J. 2006;393(Pt 1):389-96.
- 26. Njolstad PR, Sovik O, Cuesta-Munoz A, Bjorkhaug L, Massa O, Barbetti F, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588-92.
- 27. Porter JR, Shaw NJ, Barrett TG, Hattersley AT, Ellard S, Gloyn AL. Permanent neonatal diabetes in an Asian infant. J Pediatr. 2005;146:131-3.
- 28. Rubio-Cabezas O, Diaz GF, Aragones A, Argente J, Camos-Barros A. Permanent neonatal diabetes caused by a homozygous nonsense mutation in the glucokinase gene. Pediatr Diabetes. 2008;9:245-9.
- 29. Bennett K, James C, Mutair A, Al-Shaikh H, Sinani A, Hussain K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr Diabetes. 2011;12:192-6.
- 30. Massa O, Meschi F, Cuesta-Munoz A, Caumo A, Cerutti F, Toni S, et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetes Study Group of the Italian Society of Paediatric Endocrinology and Diabetes (SIEDP). Diabetologia. 2001;44(7):898-905.
- 31. Martin D, Bellanné-Chantelot C, Deschamps I, Froguel P, Robert JJ, Velho G. Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care. 2008;31(7):1321-3.
- 32. Porte D Jr, Kahn SE. Beta-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes. 2001;50(Suppl 1):S160-3.
- 33. Jeffery AN, Metcalf BS, Hosking J, Streeter AJ, Voss LD, Wilkin TJ. Age before stage: insulin resistance rises before the onset of puberty: a 9-year longitudinal study (EarlyBird 26). Diabetes Care. 2012;35(3):536-41.
- 34. Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838-49.
- 35. Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040-4.
- 36. Polak M, Dechaume A, Cavé H, Nimri R, Crosnier H, Sulmont V, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (Neonatal Diabetes) Study Group. Diabetes. 2008;57:1115-9.
- 37. Schwitzgebel VM, Mamin A, Brun T, Ritz-Laser B, Zaiko M, Maret A, et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab. 2003;88:4398-406.
- 38. deLas Heras J, Martínez R, Rica I, de Nanclares GP, Vela A, Castaño L; Spanish MODY group. Heterozygous glucokinase mutations and birth weight in Spanish children. Diabet Med. 2010;27(5):608-10.
- 39. Spyer G, Macleod KM, Shepherd M, Ellard S, Hattersley AT. Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabet Med. 2009;26(1):14-8.
- 40. Shields BM, Spyer G, Slingerland AS, Knight BA, Ellard S, Clark PM, et al. Mutations in the glucokinase gene of the fetus result in reduced placental weight. Diabetes Care. 2008;31(4):753-7.
- 41. Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet. 1998;19(3):268-70.
- 42. Velho G, Hatterlsey AT, Froguel P. Maternal diabetes alter birth weight in glucokinase-deficient (MODY2) kindred but has no influence on adult weight, height, insulin secretion or insulin sensitivity. Diabetologia. 2000;43: 1060-3.
Correspondence to:
Publication Dates
-
Publication in this collection
02 Jan 2013 -
Date of issue
Nov 2012
History
-
Received
21 June 2012 -
Accepted
10 Sept 2012