Abstract
Introduction
Hypophysitis is a chronic inflammation of the pituitary gland of complex and still incompletely defined pathogenesis. It belongs to the group of non-hormonesecreting sellar masses, sharing with them comparable clinical presentation and radiographic appearance.
Objectives
Describe the case of immunoglobulin G4 (IgG4)-related hypophysitis presenting as a mass in the sphenoid sinus.
Resumed Report
A 40-year-old Brazilian man had a diagnosis of central diabetes insipidus since 2001 associated with pituitary insufficiency. Pituitary magnetic resonance imaging revealed a centered pituitary stalk with focal nodular thickening and the presence of heterogeneous materials inside the sphenoid sinus. The patient was treated with testosterone replacement therapy. Laboratory results revealed increased IgG4 serum.
Conclusion
IgG4-related hypophysitis should be considered in patients with pituitary insufficiency associated with sellar mass and/or thickened pituitary stalk. IgG4 serum measurement for early diagnosis of IgG4-related hypophysitis should be performed.
pituitary diseases; hypopituitarism; sphenoid sinus
Introduction
Hypophysitis is a chronic inflammation of the pituitary gland of complex and still incompletely defined pathogenesis. It belongs to the group of non-hormone-secreting sellar masses, sharing with them comparable clinical presentation and radiographic appearance. These similarities often make it difficult to establish a diagnosis with certainty before pituitary surgery and pathologic examination of the resected pituitary tissue.11 Howlett TA, Levy MJ, Robertson IJ. How reliably can autoimmune hypophysitis be diagnosed without pituitary biopsy. Clin Endocrinol (Oxf) 2010;73(1):18-21 Immunoglobulin G4 (IgG4)-related diseases, including IgG4-related hypophysitis, are recently characterized entities marked by elevated serum IgG4 levels and tissue infiltration by IgG4-positive plasma cells.22 Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;366(6):539-551 33 Okazaki K, Umehara H. Are classification criteria for IgG4-RD now possible? The concept of IgG4-related disease and proposal of comprehensive diagnostic criteria in Japan. Int J Rheumatol 2012; 2012:357071 It was first diagnosed on clinical grounds in 200444 van der Vliet HJ, Perenboom RM. Multiple pseudotumors in IgG4- associated multifocal systemic fibrosis. Ann Intern Med 2004; 141(11):896-897 and then by pathology in 2007.55 Wong S, Lam WY, Wong WK, Lee KC. Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol 2007;38(11):1720-1723 We reported a patient with IgG4-related hypophysitis and summarize the current relevant literature.
Review of Literature
Hypophysitis has been classified in several ways based on anatomic location of the pituitary involvement, cause, and histopathologic appearance (Table 1).66 Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980
When surgery of the pituitary gland is performed, the pituitary pathology reveals two more common forms (lymphocytic and granulomatous) and three rarer variants (xanthomatous, necrotizing, and IgG4-producing plasma cells).66 Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980 IgG4-related hypophysitis was first reported in 2004 in a 66-year-old woman with multiple pseudotumors in the salivary glands, pancreas, and retroperitoneum,44 van der Vliet HJ, Perenboom RM. Multiple pseudotumors in IgG4- associated multifocal systemic fibrosis. Ann Intern Med 2004; 141(11):896-897 and then the entity was more extensively described in 2006 in a 70-year-old man with swelling of the salivary glands caused by a marked infiltration with lymphocytes and IgG4-positive plasma cells.77 Yamamoto M, Takahashi H, Ohara M, et al. A case of Mikulicz's disease (IgG4-related plasmacytic disease) complicated by autoimmune hypophysitis. Scand J Rheumatol 2006;35(5):410-411 Since the first IgG4-related hypophysitis case was described in 2004, more than 20 histogenetically proven cases have been reported, mostly from Japan.88 Tanabe T, Tsushima K, Yasuo M, et al. IgG4-associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern Med 2006;45(21):1243-1247 99 Isaka Y, Yoshioka K, Nishio M, et al. A case of IgG4-related multifocal fibrosclerosis complicated by central diabetes insipidus. Endocr J 2008;55(4):723-728 1010 Shimatsu A, Oki Y, Fujisawa I, Sano T. Pituitary and stalk lesions (infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: an emerging clinical entity. Endocr J 2009; 56(9):1033-1041 1111 Kotera N, Isogawa A, Uchida L, et al. [Case report: IgG4-related hypophysitis presenting with secondary adrenal insufficiency and central diabetes insipidus in a type 1 diabetes patient]. Nippon Naika Gakkai Zasshi 2011;100(4):1044-1047 All were accompanied by complications of pituitary insufficiency, but Hattori et al described the first case of IgG4-related hypophysitis without pituitary insufficiency.1212 Hattori Y, Tahara S, Ishii Y, et al. A case of IgG4-related hypophysitis without pituitary insufficiency. J Clin Endocrinol Metab 2013; 98(5):1808-1811Our patient had pituitary insufficiency.
This disease is typically part of a multifocal systemic disease recently called “IgG4-related autoimmune disease,”1313 Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003; 38(10):982-984 which emphasizes the contribution of IgG4 in establishing the diagnosis.66 Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980 IgG4, the least abundant of the four IgG antibodies, has long been associated with autoimmune and allergic diseases.1414 Kotani T, Kato E, Hirai K, Kuma K, Ohtaki S. Immunoglobulin G subclasses of anti-thyroid peroxidase autoantibodies in human autoimmune thyroid diseases. Endocrinol Jpn 1986;33(4):505-510 IgG4 antibodies were ignored for diagnostic purposes until 2001, when Hamano et al linked them to autoimmune pancreatitis and made it possible to recognize that many diseases associated with autoimmune pancreatitis share similar pathologic features, thus defining the existence of a multifocal systemic disease.1515 Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344(10):732-738 At the present time, the pathogenetic mechanism and underlying immunologic abnormalities remain unclear.1616 Umehara H, Okazaki K, Masaki Y, et al; Research Program for Intractable Disease by Ministry of Health, Labor and Welfare (MHLW) Japan G4 team. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol 2012;22(1): 1-14 A recent report identified autoimmune antibodies against GH (growth hormone) and adrenocorticotropic hormone in a patient with IgG4-related hypophysitis.1717 Landek-Salgado MA, Leporati P, Lupi I, Geis A, Caturegli P. Growth hormone and proopiomelanocortin are targeted by autoantibodies in a patient with biopsy-proven IgG4-related hypophysitis. Pituitary 2012;15(3):412-419 Nonetheless, collecting further evident cases and analysis is required to characterize the pathophysiology of IgG4-related hypophysitis.1212 Hattori Y, Tahara S, Ishii Y, et al. A case of IgG4-related hypophysitis without pituitary insufficiency. J Clin Endocrinol Metab 2013; 98(5):1808-1811
Leporati et al suggested five criteria to diagnose IgG4-related hypophysitis: (1) pituitary histopathology, (2) magnetic resonance imaging (MRI) of the pituitary, (3) biopsy-proven involvement in other organs, (4) serology with increased serum IgG4, and (5) response to glucocorticoids.66 Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980Leporati et al also proposed that the diagnosis of IgG4-related hypophysitis is established when any of the following is fulfilled: criterion 1 alone or criteria 2 + 3 or criteria 2 + 4 + 5 (Table 2).66 Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980 According to the criteria, a pituitary biopsy is not essential; however, there have been eight cases diagnosed by pituitary biopsy. Our patient fulfilled two diagnostic criteria suggested by Leporati et al to be associated with mass in the sphenoidal sinus and complication of pituitary insufficiency.
Case Report
A 40-year-old man with diagnosis of central diabetes insipidus since 2001, using oral desmopressin 0.3 mg/d, complained of frontal headache, sexual impotence, and decrease in libido. Endocrine assessment in 2003 revealed low levels of testosterone and gonadotropins and decreased insulin-like growth factor 1 (IGF-1) serum (Table 3). The patient was treated with replacement therapy of testosterone decanoate 250 mg injections every 21 days and sexual impotence and libido improved.
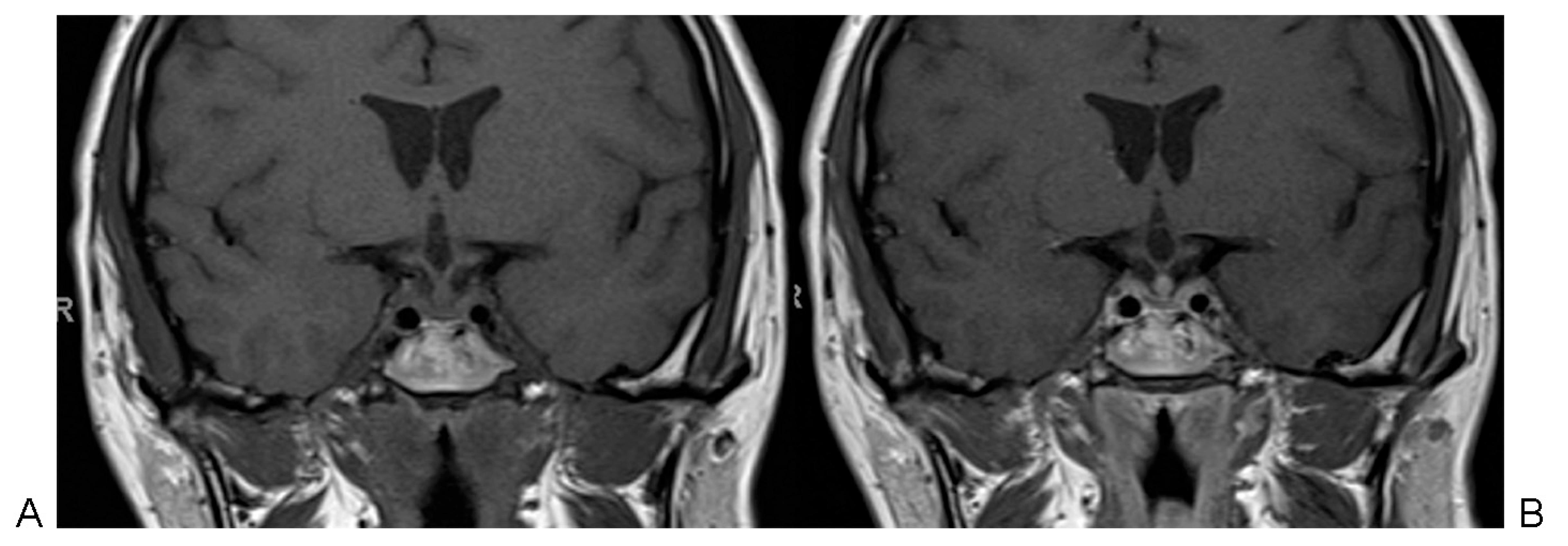
In 2005, MRI revealed a centered pituitary stalk with focal nodular thickening measuring 6 mm at the lower portion characterized by isointensity on T1-weighted images, hypointensity on T2-weighted images, and a heterogeneous intense enhancement after gadolinium administration. Heterogeneous materials inside the sphenoidal sinus were also present (Fig. 1).
Magnetic resonance imaging showing thickening of the stalk and presence of heterogeneous materials inside sphenoid sinus. (A) T1-weighted coronal image without gadolinium. (B) T1-weighted coronal image with gadolinium.
MRIs from 2008, 2010, and 2011 showed no changes on the images.
In 2012, based on the clinical and radiologic findings, a diagnosis of autoimmune hypophysitis with associated sinusitis was suspected. We observed unreactive levels of autoimmune antibodies, including anti-Sjögren syndrome antigen A (anti SS-A/Ro) and antigen B (anti SS-B/La) antibodies, ANA (antinuclear antibody) anti-Sm, rheumatoid factor, thyroglobulin, thyroperoxidase, and parietal cell. Adenosine deaminase, angiotensin I-converting enzyme, and tumor markers were negative. In 2013, there was elevation of gonadotropins, IGF-1, and total testosterone levels (Table 3). Laboratory results revealed the following: serum IgG, 1,644 mg/dL (reference range 952 to 1,538 mg/dL), and serum IgG4, 2,040 mg/dL (reference range 84 to 888 mg/dL). IgG4 was titled in two different analyses. The patient's test results strongly favored a diagnosis of IgG4-related hypophysitis.
Discussion
IgG4 serum levels are typically elevated in IgG4-related hypophysitis, similar to our patient, but they can decrease after glucocorticoid therapy initiation and in later disease stages. The involvement of the sphenoidal sinus is yet another manifestation of the multifocal IgG4-related autoimmune disease.66 Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980Our patient showed a concomitant involvement of both pituitary and sphenoidal tissues, showing that hypophysitis by IgG4 should be in the differential diagnosis of masses in the sphenoid sinus.
The typical therapy for IgG4-related hypophysitis is undefined; however, glucocorticoids are recommended as a first-line therapy against IgG4-related disease.1010 Shimatsu A, Oki Y, Fujisawa I, Sano T. Pituitary and stalk lesions (infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: an emerging clinical entity. Endocr J 2009; 56(9):1033-1041 On the basis of treatment of autoimmune pancreatitis, an initial oral prednisolone dose of 0.6 mg/kg for 2 to 4 weeks is suggested, tapered by 5 mg every 1 to 2 weeks for 2 to 3 months to determine a maintenance dose (2.5 to 5 mg/d), which should be discontinued within 3 years.1010 Shimatsu A, Oki Y, Fujisawa I, Sano T. Pituitary and stalk lesions (infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: an emerging clinical entity. Endocr J 2009; 56(9):1033-1041 1313 Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003; 38(10):982-984
Final Comments
IgG4-related hypophysitis should be considered in patients with pituitary insufficiency associated with sellar mass and/or thickened pituitary stalk. The presence of a mass in the sphenoidal sinus together with the thickening of the pituitary stalk could also suggest IgG4 hypophysitis. Furthermore, IgG4 serum measurement for early diagnosis of IgG4-related hypophysitis should be performed. Finally, a correct diagnosis is critical because it can spare the patient from a major surgery for a disease that responds well to glucocorticoids.
References
-
1Howlett TA, Levy MJ, Robertson IJ. How reliably can autoimmune hypophysitis be diagnosed without pituitary biopsy. Clin Endocrinol (Oxf) 2010;73(1):18-21
-
2Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;366(6):539-551
-
3Okazaki K, Umehara H. Are classification criteria for IgG4-RD now possible? The concept of IgG4-related disease and proposal of comprehensive diagnostic criteria in Japan. Int J Rheumatol 2012; 2012:357071
-
4van der Vliet HJ, Perenboom RM. Multiple pseudotumors in IgG4- associated multifocal systemic fibrosis. Ann Intern Med 2004; 141(11):896-897
-
5Wong S, Lam WY, Wong WK, Lee KC. Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol 2007;38(11):1720-1723
-
6Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 2011;96(7):1971-1980
-
7Yamamoto M, Takahashi H, Ohara M, et al. A case of Mikulicz's disease (IgG4-related plasmacytic disease) complicated by autoimmune hypophysitis. Scand J Rheumatol 2006;35(5):410-411
-
8Tanabe T, Tsushima K, Yasuo M, et al. IgG4-associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern Med 2006;45(21):1243-1247
-
9Isaka Y, Yoshioka K, Nishio M, et al. A case of IgG4-related multifocal fibrosclerosis complicated by central diabetes insipidus. Endocr J 2008;55(4):723-728
-
10Shimatsu A, Oki Y, Fujisawa I, Sano T. Pituitary and stalk lesions (infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: an emerging clinical entity. Endocr J 2009; 56(9):1033-1041
-
11Kotera N, Isogawa A, Uchida L, et al. [Case report: IgG4-related hypophysitis presenting with secondary adrenal insufficiency and central diabetes insipidus in a type 1 diabetes patient]. Nippon Naika Gakkai Zasshi 2011;100(4):1044-1047
-
12Hattori Y, Tahara S, Ishii Y, et al. A case of IgG4-related hypophysitis without pituitary insufficiency. J Clin Endocrinol Metab 2013; 98(5):1808-1811
-
13Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003; 38(10):982-984
-
14Kotani T, Kato E, Hirai K, Kuma K, Ohtaki S. Immunoglobulin G subclasses of anti-thyroid peroxidase autoantibodies in human autoimmune thyroid diseases. Endocrinol Jpn 1986;33(4):505-510
-
15Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344(10):732-738
-
16Umehara H, Okazaki K, Masaki Y, et al; Research Program for Intractable Disease by Ministry of Health, Labor and Welfare (MHLW) Japan G4 team. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol 2012;22(1): 1-14
-
17Landek-Salgado MA, Leporati P, Lupi I, Geis A, Caturegli P. Growth hormone and proopiomelanocortin are targeted by autoantibodies in a patient with biopsy-proven IgG4-related hypophysitis. Pituitary 2012;15(3):412-419
Publication Dates
-
Publication in this collection
July-Sept 2015
History
-
Received
03 Oct 2014 -
Accepted
24 Nov 2014