ABSTRACT
Type 2 diabetes mellitus (T2DM) is increasing in prevalence worldwide, and those non-diagnosed or misdiagnosed comprise a significant group compared to those diagnosed. Accumulated scientific evidence indicate that the current diagnostic markers (fasting glycemia, 2h glycemia after an oral glucose load and HbA1c) are indeed late diagnostic criteria when considering the incidence of diabetes-related complications and comorbidities, which are also at high risk in some groups among normoglycemic individuals. Additionally, the earlier identification of future risk of diabetes is desirable since it would allow better adherence to preventive actions such as lifestyle intervention, ultimately avoiding complications and minimizing the economic impact/burden on health care expenses. Insulin resistance and hyperhormonemia (insulin, amylin, glucagon) are non-disputable hallmarks of T2DM, which already takes place among these normoglycemic, otherwise health subjects, characterizing a state of subclinical diabetes. Insulin resistance and hyperinsulinemia can be computed from fasting plasma insulin as an independent variable in normoglycemia. An overview of the current diagnostic criteria, disease onset, complications, comorbidities and perspectives on lifestyle interventions are presented. A proposal for early detection of subclinical diabetes from routine evaluation of fasting plasma insulin, which is affordable and robust and thus applicable for the general population, is further suggested.
Key words:
diabetes; diagnosis; insulin; subclinical diabetes; metabolic syndrome
INTRODUCTION 1
According to the World Health Organization (WHO), health is defined by a complete state of well-being. However, such definition is context and clinically dependent, and not an absolute concept. The perception of apparent physical, mental and social well-being by an individual or by a health care provider is dependent upon the societal context and standards of care for diseases, which are not immutable concepts. Such reference values are typically based on population-based stratification of commonly found values and their correlation with acute markers of the onset of diseases, which might not necessarily presuppose a "complete" health state. In fact, clinical markers might vary widely among societies and ethnically, and the predisposition to the development, onset and progression of clinically diagnosed chronic diseases. Moreover, disease-related comorbidities has been found to be correlated with the stratification within normal ranges of clinical markers, raising the concern whether a chronic disease would be already in course long before the clinical diagnosis, at a subclinical stage. In light of current scientific knowledge, we discuss below whether the concept of subclinical disease would be applicable to type 2 diabetes mellitus. We further present suggestions for an earlier diagnosis of the asymptomatic pre-disease state and for the prevention of future risk of diabetes, and consequently its complications and comorbidities.
DEFINITION2
Diabetes mellitus (DM) is a cluster of chronic metabolic diseases, with many distinct characteristic although having hyperglycemia as a common marker when poorly controlled. They also have in common the malfunctioning of the endocrine pancreas, in particular the pancreatic β-cells, which are highly susceptible to varying factors such as environmental, inflammation, immunogenicity and genetic background (Schwartz et al. 2016). In type 1 DM (T1DM) insufficient production of insulin occurs due to loss in β-cells. The ADA emphasizes that T2DM is "due to a progressive loss of insulin secretion on the background of insulin resistance" (Association 2016). In fact, in type 2 DM (T2DM) the decrease in β-cell mass and insulin production is preceded by a state of hypeinsulinemia, as a compensation for an underlying state of insulin resistance (Yalow and Berson 1960, Association 1998, Garber et al. 2016) (WHO 2015). The resulting inverted-U shape pattern of the dependence of circulating insulin levels (insulinemia) on glycemia has long been known (Reaven et al. 1967, Kraft 1975), and was defined by DeFronzo as the "Starling´s curve of the pancreas" (DeFronzo 1988).
Several studies and organizations have shown an unprecedented increase in the prevalence of diabetes worldwide (NCD-RisC 2016), including T1DM (Harjutsalo et al. 2008, Patterson et al. 2009, Dabelea et al. 2014, Forga Llenas et al. 2015, Lamb et al. 2015), and the estimated number of adults with diabetes has soared to over 380 million (> 8 % world adult population, > 12% in USA) (Menke et al. 2015), with a mean estimation of over 37 % with pre-diabetes and over 45 % undiagnosed according to current diagnostic criteria (Schmidt et al. 2011, Federation 2015, Mechanick 2015, Menke 2015). The prevalence of T2DM in younger individuals has also increased worldwide (Holden et al. 2013, Menke et al. 2015), creating a new class of patients comprising children (Lustig et al. 2016), adolescents and young adults (Weiss et al. 2013, Song 2016). Such changes in phenotypic manifestation of the disease raise concern due to the life expectancy of this segment of the population and the higher cost associated with the treatment of diabetes and related complications (Huo et al. 2016). These non-adult groups in modern society are suffering from T2DM and metabolic syndrome (MetS) (clinically diagnosed by 3 out of 5 measures: elevated waist circumference, elevated triglycerides, reduced HDLc, elevated blood pressure and elevated FBG (Grundy et al. 2005), and consequently it is likely that in the short term they will form a new group of young-adult patients with life-long consequences for their own health, for their families and also for public or private health care provider. In this context, it urge the need for both earlier diagnosis of increased risk for diabetes and related complications and comorbidities (Lima 2017), requiring interventions to revert these conditions, thereby minimizing the impact on their own health and on the health care system.
CURRENT CRITERIA FOR THE DIAGNOSIS OF DIABETES 3
The diagnosis criteria of diabetes used by most diabetes societies worldwide has historically been revised in order to include the most up to date evidence correlating with the complications risk. Table I summarizes the current diabetes diagnosis criteria by the American Diabetes Association - ADA (Association 2016) and the World Health Organization - WHO (WHO 2016, Colagiuri et al. 2011), based on reference values of these clinical markers, which comprise HbA1c level over 6.5 %, a fasting plasma glucose level (FPG) over 126 mg/dL, or a plasma glucose over 200 mg/dL 2 hour after an oral load of 75g glucose (named oral glucose tolerance test, OGTT). The use of HbA1c in the diagnostic of diabetes must be regarded with caution, since several factors may affect the result, including erythrocyte lifespan (Simmons and Hlaing, 2014).
Since 2001, a new set of criteria have become commonly used for diagnosis of diabetes. According to these criteria, the overt diabetes (i.e., established, diagnosed) is preceded by two stages of non-diabetes:
-
i) pre-diabetes (also known as frank diabetes): an impaired state (impaired fasting glucose - IFG - or impaired glucose tolerance - IGT) in which these markers are altered toward the level of diabetes, and
-
ii) non-diabetic: within the normal reference range (including normoglycemia FPG < 100 mg/dL, 2h-OGTT<140 mg/mL and HbA1c < 5.7 %).
Fasting plasma insulin (FPI), c-peptide and pro-insulin are not officially adopted as clinical markers for diabetes, although recognized as possible markers of pancreatic function mostly in the form of the homeostatic model assessment (HOMA) in the estimate of β-cell function (HOMA-%B) or insulin resistance (HOMA-IR, the inverse of insulin sensitivity - HOMA-%S = 100/HOMA-IR) (Matthews et al. 1985, Wallace et al. 2004), a much simpler parameter but strongly correlated with the more complex and costly euglycemic clamp method (DeFronzo et al. 1979). Insulin, c-peptide and pro-insulin have half-life of about 5 min, 30 min and 90 min respectively (Zilker et al. 1988, Jones and Hattersley 2013). Despite the longer half-life, only about 3 % of pro-insulin remains non-cleaved, and thus limiting its use as an analytical marker. Insulin and c-peptide are produced to the same extent since c-peptide is the remaining portion of pro-insulin after cleavage. Taking into account the 6-fold longer half-life of c-peptide compared to insulin and the reduced influence of turnover due to other metabolic factors, measuring c-peptide in the assessment of insulin resistance may be advantageous either fasted of during the OGTT. In the clinical practice, insulin measurements (either FPI or during OGTT) can be prescribed and are considered accessory diagnostic tool (Wu 2006).
Despite the wide use of these established clinical markers, there has been rising concern over whether they would constitute a satisfactory panel for the clinical diagnostic of diabetes, undiagnosed diabetes and pre-diabetes or the early prediction of future diabetes and the cost-effectiveness of implementing as a general population-based approach (Zhang et al. 2005, Toscano et al. 2015).
In 1999 the DECODE group (Diabetes Epidemiology: Collaborative analysis Of Diagnostic criteria in Europe - DECODE - Study Group for the European Diabetes Epidemiology Group), with data gathered from more than 25,000 individuals with a mean follow-up of 7.3 years, that the diagnosis of established diabetes by using a FPG alone was insufficient to predict all-cause mortality, while the 2h glycemia from the OGTT provided a better prognostic information for this outcome (DECODE Study Group 1999).
In order to assess increased risk for diabetes, the ADA recommends testing to all asymptomatic individuals over 45 years old, or overweight or obese (body mass index BMI ≥ 25 kg/m2 or ≥ 23 kg/m2 in Asian descendent) with additional risk factors including physical inactivity, first-degree relative with diabetes, hypertension (≥140/90 mmHg or on therapy for hypertension) or history of CVD, HDL <35 mg/dL and/or a triglyceride level >250 mg/dL (2.82 mmol/L or on therapy for dyslipidemia), among others (Association 2016). However, by the time of their diagnosis, patients with T2DM have already an extensive (about 50 %) reduced pancreatic β-cell function (DeWitt and Hirsch, 2003), as well as signs of the prevalence of diabetes complication markers such as microalbuminuria due to kidney damage (UKPDS 1998a, Stratton et al. 2000, Bash et al. 2008) and retinopathy (Nagi et al. 1997, Kohner et al. 1998, Looker et al. 2012).
In the United Kingdom Prospective Diabetes Study (UKPDS), all microvascular and macrovascular outcomes were increasing related with HbA1c as a positive, linear continuum, with little difference for example between mean 5.6 % and 6.5 % (Stratton et al. 2000). Although not evaluated for normoglycemic non-diabetic individuals, it is unlikely that a drop to risk level equal to zero events would occur in a transition of HbA1c from 5.6 to the 5.0 % level, which suggests that even at this lower level of HbA1c a high rate of events would take place. Also in the UKPDS, the increasing severity of retinopathy was associated with lower fasting serum insulin levels (Kohner et al. 1998), which suggest a late diagnosis of diabetes, and correlates with an extensive loss of β-cell function upon diagnosis (DeWitt and Hirsch 2003).
These data suggest that the advanced complications of diabetes can be seen immediately upon diagnosis, suggesting the current "glucocentric" (Yudkin and Montori 2014) clinical criteria may constitute a late diagnosis, and thus indicating a need for earlier detection of insulin resistance and impaired pancreatic function.
RISK OF FUTURE T2DM WITHIN NORMOGLYCEMIC RANGE 4
Prospective studies have long been conducted in the search for correlations between glycemic states and the prognosis of diabetes and diabetes-related risks.
In a population-based survey of the island of Mauritius conducted with over 3,500 individuals in a follow-up of about 5 years, the incidence of diabetes was shown to progressively correlate with the FPG at baseline within the normoglycemic range (< 100 mg/dL), increasing further for individuals in FPG categories above this level, i.e., IFG (Shaw et al. 2000).
In a retrospective cohort study from the Japanese city of Omiya, conducted with over 11,000 individuals for a mean follow-up of 7 years, the risk of developing diabetes was positively correlated with the FPG at baseline irrespective of gender or age, both in the pre-diabetes range and in the normoglycemic range (Kato et al. 2009). The adjusted hazard ratio for incidence of diabetes with subjects with an FPG of 95 to 99 mg/dL was 2.3 times the risk of those with FPG < 85 mg/dL at baseline.
In an Israeli study conducted with over 13,000 men aged 26 to 45 years, with a mean follow-up of 5.7 years, a significantly augmented risk of developing T2DM was found in groups with FPG in the normoglycemic range of 87 mg/dL to 90 mg/dL (Tirosh et al. 2005). The risk or progression to T2DM was further strongly associated with higher triglyceride levels (< 150 mg/dL or > 150 mg/dL) or body mass index (BMI; <25 kg/m2, 25-29.9 kg/m2 or >30 kg/m2), both showed to be independent risk factors in this same study group.
In a large study using data from the Kaiser Permanente Northwest (KPNW) health organization in Portland, Oregon (USA) a search for correlation between normal FPG and the risk of development of T2DM was conducted (Nichols et al. 2008). Over 46,000 individuals over 40 years of age were identified with normal FPG at baseline and were followed for up to 10 years (mean follow-up time of 6.8 years) or until diagnosed diabetic. The risk of developing diabetes was positively correlated with FPG in the normal reference value (quartiles categories: < 85 mg/dL, 85 to 89 mg/dL, 90 to 94 mg/dL, 95 to 99 mg/dL) at baseline. The risk of developing diabetes was 49 % higher for those in the 90-94 mg/dL quartile compared with those with FPG below 85 mg/dL. Those subjects who developed diabetes also had some other baseline characteristic distinct from the total study sample, such as higher BMI (mean values ranging from 32.7 to 33.5 kg/m2 versus 28 to 29.9 kg/m2), higher mean triglycerides (TG; 209 to 239 mg/dL versus 142 to 164 mg/dL among the quartiles) and slightly lower mean HDLc (47 to 49 mg/dL versus 52 to 57 mg/dL among the quartiles) - which together are a characteristic of dyslipidemia. They also had hypertension (41.4 to 49.6 % versus 19.4 to 21.2 % among the quartiles) and cardiovascular disease (CVD) (8.3 to 11.9 % versus 4.8 to 7.6 % among the quartiles). These baseline characteristic of the subjects who developed diabetes shows altered values in the markers of MetS and increased risk of atherosclerosis, incidence of coronary heart disease (CHD) and mortality (Alberti et al. 2009, Wong et al. 2012). Although the risk of diabetes (4.0 %) was lower in the normoglycemic range group than in the group with pre-diabetes/IFG within the same study setting (11.3 %), both groups (normoglycemic and IFG) had close profiles among the MetS markers (BMI, TG, HDLc, high blood pressure) (Nichols et al. 2007:2). These data not only call for the importance of evaluation of both glycemic indicators, but also for the importance of the trends in MetS and insulin resistance markers. To put this into perspective, a cohort study with 56 normoglycemic individuals showed increased insulin resistance (as assessed by fasting plasma insulin - FPI - and HOMA-IR), dyslipidemia and subclinical inflammation (as evaluated by ultrasensitive C-reactive protein, hsCRP) as independent predictors of coronary artery disease as assessed by coronary artery calcium (CAC) score (Deveci et al. 2009, Budoff et al. 2013, Hecht 2015). In fact, MetS and diabetes increases the likelihood of established CAC (Wong et al. 2003), which is also suggestive of a late diagnosis when considering the risks for progression of CVD. Also, MetS and diabetes are additional risk factors in the progression of CAC (Wong et al. 2012), as shown by the Multiethnic Study of Atherosclerosis Study (MESA) conducted with over 6,810 adults individuals followed for about 4.9 years for incident CHD.
A UK retrospective study with 129 non-pregnant subjects evaluated the ability of the FPG in predicting the outcome of the OGTT (Wiener 1995). The study found that in order to achieve the lowest risk of false positive diagnoses of diabetes it would be necessary to use an upper cut-off limit of 80 mg/dL as normoglycemic range of FPG, a threshold that would rule out the need of further investigation by OGTT or any other clinical test.
Until diagnosis, diabetes can be unnoticed for over a decade under current clinical diagnosis criteria, as seen in the Whitehall II prospective occupational cohort. The participants were classified into two groups according to diagnosis at the end of the follow-up, those who developed and those who did not develop T2DM. During the study with a mean follow-up of 13 years monitored for BMI, FPG, 2h-OGTT, FPI, 2h-OGTT-insulin, HOMA-%S and HOMA-%B, those individuals which did not developed diabetes maintained a low FPG in the lower 80 mg/mL range, individuals which were ultimately diagnosed diabetic continuously increased their FPG within the normoglycemic range. Furthermore, the surge in FPG preceding diabetes diagnostic at endpoint was accompanied by increase in 2h-OGTT, decrease in HOMA-%S and in HOMA-%B, which were continuously altered since baseline throughout the follow-up trajectories. These data clearly showed a tight relation between the continuum in progression of a clinical marker within the reference normal range and the final onset of diabetes. Moreover, this study shows that the diagnosis of overt diabetes is preceded by more than a decade of asymptomatic progression in metabolic dysfunction, including decreased insulin sensitivity and β-cell function, even within their normal reference range.
The prospective Ansung-Ansan cohort study, part of the Korean Genome and Epidemiology Study, was conducted for 10 years in South Korean involving over 4100 individuals surveyed for chronic diseases. Those who progressed to diabetes and pre-diabetes at endpoint showed at baseline significantly higher BMI, waist circumference, SBP, FPG, 2h-GTT, HbA1c and HOMA-IR, and lower HOMA-%B compared to the group that did not progress, all values within their respective normal reference range (Ohn et al. 2016).
Collectively, these gathered independent studies conducted with varying ethnic groups bring evidence that the risk of future diabetes significantly increases as a function of glycemic markers and β-cell function as a continuum starting within the group of normoglycemic individuals. Below we present further data from studies correlating the risks of complications comorbidities related to diabetes in study groups in the normoglycemic range.
RISK OF DIABETES COMPLICATIONS AND COMORBIDITIES WITHIN THE NORMAL REFERENCE RANGE 5
DIABETES COMPLICATIONS IN DIABETIC INDIVIDUALS
Several studies have shown decreasing risk of diabetes complications in diabetes-diagnosed individuals that were subjected to a tight glycemic control by the use of intensive pharmaceutical therapy protocols. We refer here to two major trials, one with T1DM and other with T2DM.
The Diabetes Control and Complication Trial (DCCT) conducted with 1,441 T1DM individuals with a mean follow-up of 6.5 years showed that the intervention arm under intensive pharmaceutical therapy, with a tight glycemic control close to normal reference values, presented reduction of long-term complications such as retinopathy, nephropathy and neuropathy (DCCT 1993). The rate of progression of retinopathy fell as HbA1c decreased. These benefits observed in the DCCT showed long-term, sustained effects as observed by the lower risk of microvascular (DCCT-EDIC 2000), cardiovascular outcomes and myocardial infarction as seen in the Epidemiology of Diabetes Interventions and Complications study (EDIC), a mean follow-up of 17 years of the DCCT (Nathan et al. 2005). The UK Prospective Diabetes Study (UKPDS) study conducted with T2DM individuals followed for over 10 years showed that a tight glycemic control resulted in decrease of risk of CVD, all-cause mortality, microvascular including nephropathy (UKPDS 1998b) with sulphonylurea of insulin, and macrovascular diseases (myocardial infarction, sudden death, angina, stroke, peripheral disease) with metformin (UKPDS 1998a).
DIABETES COMPLICATIONS ON NON-DIABETIC INDIVIDUALS
The risks of complications of diabetes (such as neuropathy, nephropathy, retinopathy, CVD, vascular, among others) have also been observed as a function of metabolic dysfunction among normoglycemic individuals.
Retinopathy
The trend in increasing of the prevalence of any retinopathy has been seen as a HbA1c dependent variable within the normal reference range in cross-sectional epidemiological studies with the Pima Indians, an Egyptian study, and the third National Health and Nutrition Examination Survey (NHANES) (WHO 2015, Colagiuri et al. 2011).
Cardiovascular disease
In the US San Antonio Heart Study, over 2,560 Mexican-American and non-Hispanic individuals non-diabetic at baseline were followed for 8 years. The risk of CVD was evaluated and the HOMA-IR and FPI were found to be independent risk factor with FPG and 2h-OGTT within the normoglycemic range (Hanley et al. 2002).
A diagnosis of T2DM has been considered an independent risk factor for CVD (Valenti et al. 2016). The prospective DECODE study comprising European 22-cohorts involving over 29,700 individuals in a median follow-up of 11 years reported an increasing risk of all-cause CVD and non-CVD mortality for individuals with 2h-OGTT levels within the normoglycemic range (below 140 mg/dL or 7.7 mM) (DECODE Study Group, European Diabetes Epidemiology Group 2003)
In a longitudinal population-based prospective study conducted in the Japanese town of Hisayama, a total of 2,851 individuals were followed for about 7 years and CVD were found to correlate with HbA1c within the non-diabetic range (Ikeda et al. 2013).
In a 10-year follow-up of the Dutch Hoorn cohort study involving 1,647 non-diabetic subjects (eligible for final data analysis), HbA1c was shown to be an independent predictor of CVD (Van't Riet et al. 2012)
In the Italian Bruneck study conducted with over 910 adults followed for 15 years, the incidence of CVD was increasingly higher in the upper quartiles of HOMA-IR or FPI at baseline even after adjustment for varying risk factors (Bonora et al. 2007).
In the Australian Diabetes, Obesity and Lifestyle (AusDiab) study conducted with over 10,000 subjects over 25 years of age in a follow-up of 7 years, the hazards for all-cause mortality and CVD mortality were independently correlated with 2h-OGTT and HbA1c even within their normal reference range (Barr et al. 2009). Also in the AusDiab cohort, conducted with general non-diabetic population and involving over 8,530 adults over 35 years of age followed for about 5 years the incidence of fatal or non-fatal CVD showed a progression correlated with HOMA-%S, with a hazard ratio of about 2.0 when comparing the first and fifth quintiles (Barr et al. 2010).
In a UK-based prospective population study in which over 10,000 adults were followed for an average of 6 years, an increased risk for fatal and non-fatal coronary heart disease (CHD) cardiovascular events and total mortality associated was found with increasing HbA1c levels in the normal reference range (Khaw et al. 2004).
In the Atherosclerosis Risk in Communities (ARIC) Study, several evidences of diabetes complications have been associated with HbA1c in the normal, non-diabetic reference range (< 5.7 %). In an ARIC prospective case-cohort study conducted for over 8 years of follow-up with over 1,320 non-diabetic adults, a progressive increase in risk of CHD was found as a function of HbA1c in the normal reference range among those in the normoglycemic range (NCD-RisC 2016). Another ARIC study with over 10,880 non-diabetic individuals in a mean follow-up of 8 to 10 years found a correlation of HbA1c within normal reference range with increasing risk of incident ischaemic stroke (Selvin et al. 2005b). A median follow-up of 14 years conducted with over 11,090 non-diabetic adults found a positive correlation between the incidence rate of new diagnosed diabetes, CHD and stroke with HbA1c in its normal range.(Selvin et al. 2010)
In a Canadian prospective study involving over 2100 non-diabetic adult men, the risk of ischemic heart diseases (IHD) was shown to be independently correlated with fasting hyperinsulinemia compared to matched patients with no occurrence of IHD within the 5 years of follow-up (Després et al. 1996). Although the risk of IHD was independent from dyslipidemic state, it showed a synergic effect with increasing triglyceride. In another population-based cohort study (NHANES III) involving over 13,100 non-diabetic individuals over 20 years, c-peptide was found positively correlated with increasing risk of CVD deaths in nondiabetic patients and with decreasing levels of serum HDLc (Li et al. 2015). These studies did not exclude pre-diabetic groups. Although well-known that the risks of CVD is increased in pre-diabetic and diabetic compared to non-diabetic population (Stratton et al. 2000), the risk of first time stroke among normoglycemic Swedish population was positively correlated with fasting pro-insulin and insulin levels, even more pronounced in women (mean OR 13.7) than in men (mean OR 3.4) (Lindahl et al. 2000).
The Spanish cohort from the Canary Islands conducted with over 6,600 adults from the general population showed elevated c-peptide fasting levels associated with increasing risk of coronary disease - acute myocardial infarction (AMI) and coronary artery disease (CAD) - within normoglycemic individuals (Cabrera de León et al. 2015), which also correlated positively with increasing obesity, TG hypertension and HOMA-IR, and lower HDLc.
In a cohort study with 1,073 non-diabetic adult patients in the normoglycemic range (for FPG, HbA1c) which survived AMI, they found an independent association of incident multivessel coronary artery disease (CAD) with HOMA-IR (Karrowni et al. 2013). In this same cohort, the quantiles of HOMA-IR was statistically and positively correlated with increasing FPG, FPI, HbA1c, hsCRP, TG, and BMI.
Cancer
In the Japan Public Health Center-based prospective study (JPHC Study Group) which followed over 29,000 individuals which were cancer-free at baseline. And after a median follow-up of 8.5 years, they found increasing risk of cancer in individuals with higher HbA1c levels within both non-diabetic and diabetic ranges (Goto et al. 2016).
In an Italian prospective case controlled study, involving over 10,000 women followed for over 5 years, FPG within the normal reference value of non-diabetic was found associated with increasing risk of breast cancer (Muti et al. 2002, Sieri et al. 2012).
Dementia
During a median follow-up of 6.8 years of over 2,000 patients, the Adult Changes in Thought (ACT) study found a progressive increasing risk for development and diagnosis of dementia as a function of glycemia at baseline in groups without diabetes (Crane et al. 2013), suggesting insulin resistance and increased microvascular complications in the central nervous system as underlying causes.
In a US cohort from northern Manhattan involving adults over 65 years of age and no sign of dementia at baseline, the odds of Alzheimer disease (AD) and dementia was increased over 100 % in individuals with hyperinsulinemia (Luchsinger et al. 2004).
Renal diseases
In the Atherosclerosis Risk in Communities (ARIC) Study, a community-based cohort with over 1,800 diabetic participants, a positive association between chronic kidney disease (CKD) - in the absence of albuminuria and retinopathy - and a broad HbA1c concentration range, including the normal reference values (<5.7 %)
In conjunction, these data reported above, among others studies reported elsewhere, illustrate that the risks of incidence of diabetes-related complications and comorbidities correlate with markers of glucose metabolism in a dose-dependent manner within the normal reference range for non-diabetic. Consequently, these data indicate the existence of initial manifestation of diabetes-related complications in subclinical conditions not detected by the current diagnostic approach. The missing link relies on the identity of one or more causal agents, or even the identification of a common independent risk factor that would better describe the increased risk of progression to diabetes and related complications and comorbidities among normoglycemic individuals. The clinical markers FPG, OGTT and HbA1c are glycemic proxies highly influenced by a large subset of variables (Rydén et al. 2016), including insulin secretion and insulin resistance, glucagon control, glucose absorption (during the GTT), cortisol, stress hormones, among others. Among these factors, insulin secretion - along with amylin cosecretion - have been shown to be regulated accordingly in order to keep the glucose homeostasis, and thus pancreatic β-cell function is a valuable tool to be investigated.
SUBCLINICAL DIABETES DIAGNOSIS: HYPERHORMONEMIA AND INSULIN RESISTANCE AS THE MISSING PROXIES 6
After diagnosis, the progression of T2DM is well characterized by the decline in pancreatic β-cell function (Butler et al. 2003, DeWitt and Hirsch 2003, Association 2016). However, the decrease in β-cell function and insulin secretion with the progression of both frank and overt diabetes is preceded by a hyperhormonemia state, in which hormonal (both insulin and amylin) secretion by the β-cells attempts to offset the glucose load and the varying degree of insulin resistance, keeping glycemia in the normoglycemic range (Abdul-Ghani and DeFronzo 2009). Such observation makes the case for pancreatic hyperhormonemia as a major ontogenic hallmark of diabetes. It follows that evaluation of hormonal levels become a natural candidate as a more specific and sensitive clinical diagnostic proxy in normoglycemic individuals, otherwise healthy and non-diabetic according to the current glucocentric diagnostic approach.
FROM THE BIHORMONAL HYPOTHESIS TO THE ISLET MULTIHORMONAL DISFUNCTION
Glucagon
Glucagon is an hormone secreted by the α-cells from pancreatic islets with varying systemic physiologic roles including the maintenance of normoglycemia under fasting, as long recognized by evidences from Unger, Cahill and colleagues (Marliss et al. 1970). The evidences that glucagon levels in individuals with overt diabetes were elevated lead the proposal of the "bihormonal hypothesis" of T2DM in 1975 by Unger and Orci (Dobbs et al. 1975, Unger and Orci 1975), by which hyperglycemia was explained by the lack of insulin regulation of glucose metabolism and paracrine antagonization of excessive secretion of glucagon, currently recognized as a key strategy in the therapeutics of diabetes (Godoy-Matos 2014, Hay et al. 2015, Sisnande et al. 2015, Bower and Hay 2016). In fact, reduced insulin secretion and acute glucagon secretion upon stimulation with arginine is observed in IGT individuals years before diagnosis, as found by Ahrén and colleagues (Larsson et al. 1995, Ahrén 2009). Such traits are found in subjects that were normoglycemic and normoglucagonemic at baseline, but already showing higher FBI at baseline within normal reference range.
Pancreatic Polypeptide (PP)
The pancreatic polypeptide (PP) is secreted by the PP cells and has long been shown to be elevated in T2DM individuals (Floyd et al. 1976). In fact, during an OGTT the insulin levels in pre-diabetic are higher than in non-diabetic individuals, while PP levels and glucagon are similar between these groups. However, in T2DM subjects both insulin, PP and glucagon levels are elevated during an OGTT (Chia et al. 2014), bringing evidences for a further multihormonal hypothesis.
Amylin
Amylin is a 37 amino acid hormone belonging to the calcitonin gene-related peptide (CGRP) family discovered in 1987 (Cooper et al. 1987, Westermark et al. 1987) and is concomitantly released along with insulin by the same secretory granules of the pancreatic β-cells (Guerreiro et al. 2013). Among the physiologic functions, amylin has a central role in the regulation of glucagon secretion (Young, 2005). During OGTT in non-diabetic (30 min OGTT peak <145 mg/dL, 1h OGTT < ~ 130 mg/dL, 2h OGTT < ~90 mg/dL) obese adults, both insulin (1h peak ~ 57 mU/L, ~400 pM) and amylin (1h peak ~ 8 pM) are cosecreted (Thomaseth et al. 1997). They are secreted at an apparent constant ratio in non-diabetic individuals, reaching a serum concentration ratio at 1 h peak of about 50mol: 1mol insulin:amylin. In normoglycemic individuals progressing toward pancreatic impairment, a parallel hyperhormonemia (hyperinsulinemia and hyperamylinemia) follows, with an increase in the relative amount of amylin to insulin as a function of progression from NGT to IGT and finally T2DM (Ludvik et al. 1991, Thomaseth et al. 1997, Kahn et al. 1998).
From this islet hormonal panel we notice that insulin and amylin are hormone that may be found elevated in fasting state as well as after an oral glucose load in some normoglycemic non-diabetic individuals, while glucagon and PP are more likely to be altered in diagnosed diabetic subjects.
Insulin
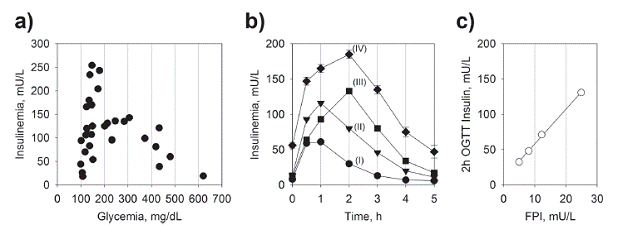
Yalow, Berson and colleagues have reported a large variability in plasma insulin concentration in normoglycemic patients undergoing OGTT (Yalow and Berson 1960, Yalow et al. 1965), also corroborated by subsequent study by Reaven and colleagues (Reaven et al. 1969). In another study with individuals undergoing GTT, Reaven and colleagues have further found a biphasic dependence of insulinemic response on glycemia, at first reporting a steep increase within the normoglycemic range which preceded a descent phase as a function of increasing glycemia within the IGT and diabetic ranges (Reaven et al. 1967) (Fig. 1a). Kraft has further confirmed the hyperinsulinemic response to an oral glucose load during an extended OGTT - up to 5 hours - among normoglycemic individuals, which anticipates the decline in insulin response observed in the overt diabetes (Kraft 1975, 2008) (Fig. 1b). The varying degrees of insulinemic responses during OGTT were stratified into quintiles (so-called "Pattern") and correlate with the levels of FPI (Kraft 2008). These varying levels of insulinemic response to OGTT - or even meals - among those otherwise considered healthy individuals, provides evidence for varying degrees of insulin resistance among normoglycemic subjects. These data have also been repeatedly observed by other groups.

Insulinemic response following an oral glucose load. a) Insulinemic response in individuals following an oral load of glucose. Data from (Reaven et al. 1967). b) Insulinemic response in normoglycemic individuals following an oral glucose load. Each curves (I, II, III and IV) represent a Pattern (quantile) with average of the insulinemic responses from a large subset of individuals following an oral glucose load. Data from (Kraft 2008). c) Correlation between FPI and 2h insulinemia following an oral glucose load. Data form (Johnson et al. 2010).
When comparing groups of normoglycemic non-obese and obese adult individuals, the latter shows both an increased FPI and 24-h insulinemic response, with minor difference in the 24-h glycemic profile (Polonsky et al. 1988). In a study with normoglycemic obese (BMI ~ 35 kg/cm2) Latino and Afro-American youth, the group was hyperinsulinemic (FPI > 27 U/mL), and consequently insulin resistant (HOMA-OR > 4.3) and also an IGT with high insulinemic profile during OGTT (Lustig et al. 2016). However, even among lean, normoglycemic and normolipidemic (fasting) young individuals a subgroup with elevated FPI can be found - yet "normal" (< 13 mU/mL) - showing a hyperinsulinemic and dyslipidemic 24h profile - in particular high TG and de-novo hepatic lipogenesis (Petersen et al. 2007). In another study, Finnish individuals were screened for their degrees of insulin resistance using euglycemic hyperinsulinemic clamp, and the results were compared with their FPI (Laakso 1993). In the group with NGT, the prevalence of insulin resistance increased as a function of their FPI, matching similar prevalence of those in the frank (IGT) or overt (diagnosed) diabetic groups at a FPI of 13 mU/L and above. In the San Antonio Metabolism (SAM) study conducted with 388 obese and non-obese subjects (NGT, IGT and T2DM), the insulin secretion during OGTT showed a typical inverted-U shape when plotted as a function of FPG, showing the decline in insulin secretion preceded by an hypersinsulinemic phase in the normoglycemic range (Gastaldelli et al. 2004). This study also found that the insulin secretion along the OGTT correlated with the degree of insulin resistance as assessed by euglycemic insulin clamp. It also found that the decline in β-cell function is an exponential continuum as a function of FPG (or 2h-OGTT), beginning in the normoglycemic range. In a retrospective US study a group of over 675 subject with FPG in the normoglycemic range, FPI and HOMA-IR positively correlated with 2h-OGTT insulin (Johnson et al. 2010) (Fig. 1c).
INSULIN RESISTANCE, HYPERINSULINEMIA AND HEPATIC MANIFESTATION OF METABOLIC SYNDROME
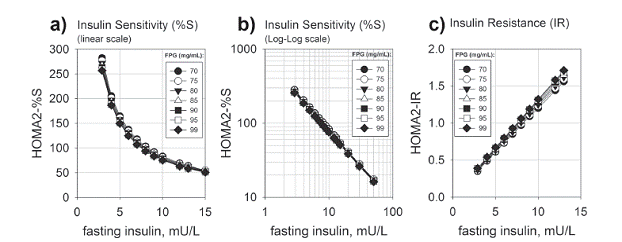
We recall that HOMA is a measure calculated from results of measurements of fasting glycemia and insulinemia during routine blood work. HOMA is well accepted due to its simplicity and an analytic power comparable to clamps, golden standards for evaluation of insulin resistance but a complex test not suitable for routine clinical diagnostic. Although well known as a suitable proxy for evaluation of β-cell function and insulin resistance in individuals diagnosed with frank or overt diabetes, HOMA can also be a direct evidence for β-cell function in the normal reference range, and thus an appropriate - and affordable - proxy. In order to keep a tight glycemic control, individuals in the normoglycemic range show increasing insulin resistance, which manifests itself through increasing insulinemia both fasting and in response to an oral glycemic load. This increasing insulin resistance is phenotypically manifested as an increasing insulinemia, which is reflected as a steep decline in HOMA-%S (Figs. 2a, b) and a linear increase in HOMA-IR (Fig. 2c). According to this model, it is noted that an increase from 2 mU/L to about 15 mU/L in FPI is followed by an 8 fold decline in HOMA-%S, from 400 to 50 (as interpolated from Fig. 2b). These modeled data are confirmed by experimental results, showing the extensive increase in prevalence of insulin resistance within the normal reference range of FPI (< 13 mU/L) and normoglycemic range (Laakso 1993).

Insulin sensitivity as a function of fasting plasma insulin according to HOMA2 model. The HOMA2 model as implemented in the HOMA calculator (https://www.dtu.ox.ac.uk/homacalculator/) (Wallace et al. 2004) was used to diagram the insulin sensitivity (HOMA2-%S, the inverse of insulin resistance) and insulin resistance (HOMA2-IR) as a function of fasting plasma insulin. a) HOMA2-%S, linear scale; b) HOMA2-%S, log-log scale. c) HOMA2-IR. Numbers in the legends correspond to the FPG levels used in simulation, within the normoglycemic range (from 70 to 99 mg/L). Notice that insulin sensitivity shows a steep dependence on the fasting insulin (a) behaving as a double exponential decay function and thus linear in log-log scale (b). Interpolation of this data allows the observation of a decrease in half of insulin sensitivity when FPI rises from 2 to 5 mU/L FPI, and a further decrease in half from 5 to 10 mU/L. Even within the normal reference range of FPI (up to 13 mU/L), a shift from 2 to 10 mU/L in FPI corresponds to a decrease to about 25 % of initial insulin sensitivity, a clear sign of advanced insulin resistance (c) and thus subclinical diabetes in the normoglycemic range.
From decades of independent studies it becomes evident that an early multihormonal disfunction takes place in as an attempt to control glycemia in the context of the metabolic degeneration. More importantly, this analysis shows that FPI is a direct and reliable indicator of insulin resistance in otherwise healthy non-diabetic normoglycemic individuals. In fact, the importance for the assay of FPI in the evaluation of insulin resistance before the onset of the clinical diabetes has long been recognized by the ADA for almost 20 years, as stated in their official statement "Consensus Development Conference on Insulin Resistance" (Association 1998). In that report, the non-recommendation for the routine screening of fasting insulin relied for the most part in analytical limitations, in the absence of clear cut-point criteria, and in the lack of correlational evidence between insulin resistance and outcomes.
The present body of evidence indicates that the glucocentric approach (FPG, HbA1c and OGTT) does not conclusively predict future risk of diabetes and related complications/comorbidities. Given that insulin resistance is the underlying basis for glucose intolerance leading to rise in glycemia and all others diagnostic markers for diabetes and pre-diabetes and that insulin resistance may builds up even in normoglycemic individuals, the direct assessment of insulin resistance should constitute the diagnostic panel in the screening of diabetes, requiring solely a FPI and/or a confirmatory OGTT with insulin measurement.
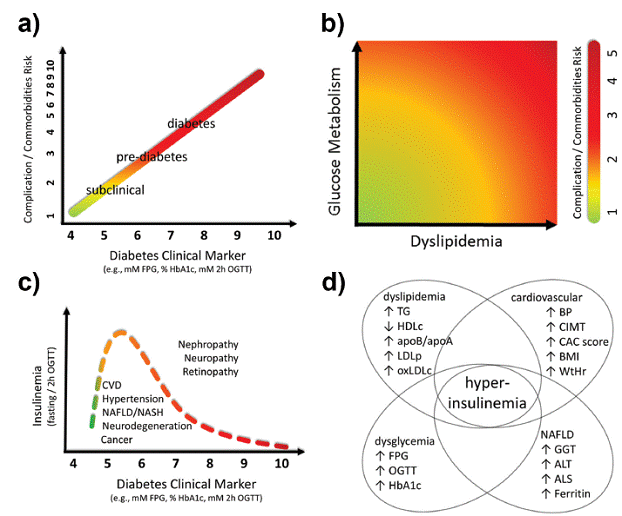
The insulin resistance index (HOMA-IR) by means of FPI may be evaluated along with a broader metabolic panel prospecting the intraorgan origin of insulin resistance, in particular hepatic by means of gamma-glutamyl transferase (GGT), alanine aminotransferase (ALT), aspartate aminotransferase (AST) (Unwin et al. 2015, Oniki et al. 2016) and ferritin (Brudevold et al. 2008). Non-alcoholic fatty liver disease (NAFLD) spectrum has been considered an hepatic manifestation and/or precursor of MetS (Smits et al. 2013, Lonardo et al. 2015) and even elevated levels within the normal reference range have been associated with markers of MetS, NAFLD and T2DM (Sanyal et al. 2015), and thus a subclinical stage of MetS. Furthermore, other anthropometric markers associated to MetS should be evaluated since accumulation of MetS markers are indicative of increased insulin resistance (Garg et al. 2011). These MetS markers include subcutaneous, abdominal and intravisceral fat (BMI, waist to hip ratio), cardiovascular and atherosclerotic outcomes (such as carotid intima-media thickness - CIMT (Pais et al. 2016), coronary artery calcium - CAC, also known as calcium score (Valenti et al. 2015)), blood pressure, inflammation (hsCRP) and atherogenic dyslipidemia - high circulating triglycerides, low HDLc, high apolipoprotein B or high LDL particle number (Leroux et al. 2000, Pourfarzib et al. 2014), high small LDL particles and oxidized LDLc (Boizel et al. 2000), some of them assessed as the lipoprotein insulin resistance index - LPIR score, which shows strong association with HOMA-IR (Shalaurova et al. 2014) (Fig. 3).

Schematic diagram of the continuum of risk of common diabetes complications as a function of glucose metabolism markers. A continuous color gradient is depicted ranging from lower (green, lower left side) to higher risks (red, upper right side). a) Schematic representation of the progression of risks (log scale) of future complications and/or comorbidities related to currently adopted official diabetes clinical markers (FPG and/or HbA1c and/or 2h-OGTT) and at normoglycemic range (subclinical diabetes). Diagram inspired on the UKPDS (Stratton et al. 2000) and ARCS (Selvin et al. 2005a) studies. b) The progression of complications / comorbidities of diabetes in the subclinical range (within the normal reference ranges, without established diabetes symptoms) of glycemic and lipid markers. Glucose metabolism state for FPG, FPI/c-peptide/pro-insulin, HbA1c, 2h-OGTT glucose. Dyslipidemia states for TG, HDL, TG/HDL ratio, oxLDL. Diagram inspired on the study of (Tirosh et al. 2005). c) Schematic representation of the biphasic behavior of insulin secretion as a function of progression of diabetes. At low, normoglycemic reference values of glucose indicators, insulin secretion is increased in order to compensate for building insulin resistance. Diagram inspired by Reaven (Reaven et al. 1967), Kraft (Kraft 1975), and DeFronzo (Gastaldelli et al. 2004) studies. d) Schematic representation of the clinical markers associated with hyperinsulinemia within normal reference ranges.
INTERVENTION IN THE PREVENTION OF DIABETES7,8
We present below a brief perspective on some of them to shed light on the manageable aspect of the metabolic dysfunction by means of lifestyle interventions.
MEDITERRANEAN DIET APPROACH
Lifestyle intervention showed effective in the prevention of T2DM in non-diabetic individuals at high risk of CVD who followed a Mediterranean diet approach (high in vegetables, olive oil, fish, nuts, fiber, polyphenos, α-linoleic acid, and minimally processed in natura products, low linoleic acid and ultraprocessed food items) for a mean follow-up of 5 years (Salas-Salvadó et al. 2011, Estruch et al. 2013, 2016, Babio et al. 2014).
The Diabetes Prevention Program (DPP) study (DPP 2002) conducted with high risk group showed a 58 % reduction in the risk of diabetes by lifestyle intervention, which included the National Cholesterol Education Program Step 1 (NCEP Step 1) diet, similar to a Mediterranean Diet (Kris-Etherton et al. 2001). The lifestyle intervention in the DPP proved to be superior to metformin (850 mg b.i.d.) intervention, both compared to placebo.
CALORIC RESTRICTION
Another approach in the control of diabetes markers is caloric restriction. This approach has been used for over 100 years, long before the availability of insulin and other drugs for diabetes (Mazur 2011). Modern studies have been confirming the caloric restriction protocol and the reversal of organ dysfunction and the reversal of diabetes markers (Steven et al. 2016) and has been also recognized by the ADA as an intervention in the improvement of insulin resistance (Association 1998). According to this protocol, not only is total caloric intake decreased, but also the glycemic load, which introduces confounders, making it difficult to extrapolate the individual benefits of each component in a multivariate analysis.
Although some authors argue that isoenergetic meals varying in content of macronutrient may behave similarly in the short term (Hall et al. 2015), metabolic markers and the long term impact of a large set of variables influences considerably the metabolism according to the macronutrient distribution (Gardner et al. 2007), insulin resistance status (Ebbeling et al. 2007, McClain et al. 2013), the content of resistant starch (Leeman et al. 2005) and the use of seasoning components such as vinegar (Ostman et al. 2005), fats as olive oil and butter (Bozzetto et al. 2016), among others variables. Given that the post-prandial insulin spike may take about 5 h to reach the baseline level (Gannon and Nuttall, 2004), the meal distribution over the course of the day should be taken into account since it is likely to impact the 24 h insulinemic profile (Kahleova et al. 2014).
It is also recognized the existence of large population variability in glycemic response to similar meals (Zeevi et al. 2015), the intra-individual variability in glycemic response to different meals with equivalent macronutrient load (e.g., carbohydrate: starch + sugar) (Hätönen et al. 2006), which poses further uncertainty on the adequacy of total caloric restriction protocols, the validity of the calorie-in-calorie-out model (Hall et al. 2015), and the usefulness of the glycemic index (Jenkins et al. 1981, Atkinson et al. 2008) model.
CARBOHYDRATE /GLUCOSE LOAD RESTRICTION
Taking into account the needs for decreasing in the risk of future diabetes and complications thereof, a lifestyle interventional approach that decreases endogenous insulin requirements, improve insulin sensitivity, pancreatic function and improves evident risk markers such as TG, HDL, insulinemia and glycemia should be considered.
A direct correlation between glycemic load from dietary carbohydrate and physiologic insulin requirement - currently known as food insulin index (FII) - is well-established. It is well-known that glucose load induces expressive increase in glycemia and insulin, while other macronutrients loads do not significantly change both markers (Robertson et al. 2002, Bao et al. 2009). Prioritization of meal preparations with low FII and thus with low bioavailable glucose from total carbohydrate contents such as starch and sugars along with the preferred use non ultra-processed food items (Canella et al. 2014) is likely to compose a strategy to decrease the excessive demand upon pancreatic function, to overcome long-term hyperinsulinemic status, reduction in insulin resistance and in the likely of development of MetS-related outcomes.
For several decades now, the role of glycemic load in the long term rise of triglycerides has been known, and it is worsened according to the individual insulin responses (Reaven et al. 1967). The hypertriglyceridemic effect of fructose from sugars and its impact on markers of MetS is also well documented (Lim et al. 2010), and its dietary reduction has been shown to be associated with improvement in MetS markers (Schwarz et al. 2015, Lustig et al. 2016)
Moreover, it has been postulated that most metabolic diseases show increased risk under an hyperinsulinemic condition (Cordain et al. 2003, Kopp 2003). In fact, a study with over 4,150 participants from Northern China followed for over 4.2 years suggested that even when participants in a cohort were normalized for similar baseline characteristics (BMI, age, age, smoking, waist circumference - lower limit of 81 cm - SBP, DBP, normoglycemic range (about 4.8 mM = 87 mg/dL), fasting plasma insulin (about 7 mU/L), total cholesterol, HDLc (about 50 mg/dL), obesity, hypertension and hyperlipidemia, but with SBP and triglyceride (in the upper normal range),the study found correlation of increasing consumption of starchy foods with the probability of MetS, indicating the existence of the disease and their probable complications and comorbidities.
The average load of essential micronutrients expressed as percentage of the dietary reference intake (DRI) show a positive correlation with the overall content of protein in unprocessed or minimally processed food items (Fig. 4a), while displaying a lower prevalence in starch- and sugar- rich food items (Fig. 4b and Fig. 4c, respectively), which are the main origin of exogenous glucose (USDA 2014). Limiting the intake of essential micronutrients can pose an increasing risk for the prevalence of diabetes and other metabolic disorders (Ames 2006, Miao et al. 2013, Kaur and Henry 2014). Given that the insulin demand is directly related to exogenous glucose (Bao et al. 2011), decreasing the consumption of starch and sugar - rich food items is a better strategy for the control of both established diabetes and prediabetes (Feinman et al. 2015, Tóth and Clemens 2015) as well as subclinical diabetes (Lima 2017). There is no minimum human requirement for exogenous glucose since the human body can produce glucose (Institute of Medicine 2005, Cahill 2006) and use it along with ketone bodies as main energetic source in many organs including brain (Cunnane and Crawford 2003, Institute of Medicine 2006), provided adequate amounts of nutrient-rich protein and good-quality lipids are consumed.

Correlation between the average content of essential micronutrients and macronutrients load. The average load of 28 essential micronutrients in non-processed food items (",raw" keyword filter, resulting in 1.325 food items) was calculated from the whole USDA database (SR27) in the basis of % of the Dietary Reference Intake (DRI; males, 31-50 years old) for each 100 g product and reported here. As a quick reference for high micronutrient load, consider the distribution above 50 % DRI. Essential amino acids were not included in order to avoid bias by skewing data toward high micronutrient in protein. a) Ternary plot representing the proportion of macronutrient (PTN = protein, CHO = carbohydrate, FAT = lipids) as % energy. Each symbol represent one non-processed (",raw") food item and the size is proportional to the average load of the 28 essential micronutrients expressed as % of the DRI. b) Average load of 28 essential micronutrients (% DRI, male, 31-50 years old) in 100 g unprocessed food item as a function of sugar content. c) Average load of 28 essential micronutrients (% DRI, male, 31-50 years old) in 100 g unprocessed food item as a function of starch content. Notice the LOG-LOG scale. Data from USDA National Nutrient Database for Standard Reference, Release 27 (USDA, 2014:27) (SR27; http://www.ars.usda.gov/) and Dietary Reference Intake (DRI) for adult male 31-50 years-old (http://ods.od.nih.gov/Health_Information/Dietary_Reference_Intakes.aspx). Micronutrients: Ca, Fe, Mg, P, K, Na, Zn, Cu, F, Mn, Se, VitA, VitE, Vit D (D2 + D3), Vit. C, Thiamin, Riboflavin, Niacin, Pantothenic Acid, Vit B6, Folate (total), Vit. B12, Choline (total), Vit. K (phylloquinone), Folate (food), Linoleic Acid (LA), α-linolenic acid, and also included EPA+DHA (not in sr27, but known for its limited endogenous production and requirement from exogenous source (Mozaffarian and Wu 2012).
CONCLUSIONS 9
There has been intense debate whether the current thresholds for diagnosis of diabetes should be decreased and if HbA1c and other additional metrics could compose a more complete risk assessments panel for diabetes and its complications (Cefalu 2016, Yudkin 2016). The extensive scientific literature cited above displays sound evidence for the strict dependence of markers of metabolic disorder as a continuum of insulinemia and as a direct indicator of the trajectory of insulin resistance taking place among normoglycemic individuals.
It is time to reappraise the diagnosis criteria beyond the sole use of the glucocentric markers, and to evaluate the levels of insulin resistance (through insulinemia) and hepatic manifestation of metabolic syndrome (by means of lipid metabolism, even within the normolipidemic range but at outer percentiles), which comprise a set of cost-accessible diagnostic parameters of direct markers of the underlying basis of the disease. Under the patient perspective, it would allow the health care provider to propose early non-pharmacological lifestyle intervention once identified a subclinical diabetes pattern, aiming to improve overall health and reducing the risks of future diabetes and the evident risks of diabetes-related complication and comorbidities. From the public health perspective, adopting policies aimed at the promotion of a healthy nutrition and lifestyle and the consequent minimization of the burden of diabetes, complications and comorbidities would reduce the economic impact on the health care system and society overall.
ACKNOWLEDGMENTS
The author would like to thank Mr. Alexander Ruhle for critical review of the manuscript. This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro Carlos Chagas Filho (FAPERJ). The funding agencies had no role in the study design, data collection and analysis, or decision to publish or prepare of the manuscript. Prof. Luis Mauricio T. R. Lima is applicant of patents regarding controlled release of peptides.
REFERENCES
- ABDUL-GHANI MA and RA DEFRONZO. 2009. Plasma glucose concentration and prediction of future risk of type 2 diabetes. Diabetes Care 32(Suppl 2): S194-198.
- AHRÉN B. 2009. Beta- and alpha-cell dysfunction in subjects developing impaired glucose tolerance: outcome of a 12-year prospective study in postmenopausal Caucasian women. Diabetes 58: 726-731.
- ALBERTI KGMM ET AL. 2009. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120: 1640-1645.
- AMES BN. 2006. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc Natl Acad Sci U S A 103: 17589-17594.
- ASSOCIATION AD. 1998. Consensus Development Conference on Insulin Resistance: 5-6 November 1997. Diabetes Care 21: 310-314.
- ASSOCIATION AD. 2014. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 37: S81-S90.
- ASSOCIATION AD. 2016. 2. Classification and Diagnosis of Diabetes. Diabetes Care 39: S13-S22.
- ATKINSON FS, FOSTER-POWELL K and BRAND-MILLER JC. 2008. International Tables of Glycemic Index and Glycemic Load Values: 2008. Diabetes Care 31: 2281-2283.
- BABIO N ET AL. 2014. Mediterranean diets and metabolic syndrome status in the PREDIMED randomized trial. CMAJ 186: E649-657.
- BAO J, ATKINSON F, PETOCZ P, WILLETT WC and BRAND-MILLER JC. 2011. Prediction of postprandial glycemia and insulinemia in lean, young, healthy adults: glycemic load compared with carbohydrate content alone. Am J Clin Nutr 93: 984-996.
- BAO J, DE JONG V, ATKINSON F, PETOCZ P and BRAND-MILLER JC. 2009. Food insulin index: physiologic basis for predicting insulin demand evoked by composite meals. Am J Clin Nutr 90: 986-992.
- BARR ELM, BOYKO EJ, ZIMMET PZ, WOLFE R, TONKIN AM and SHAW JE. 2009. Continuous relationships between non-diabetic hyperglycaemia and both cardiovascular disease and all-cause mortality: the Australian Diabetes, Obesity, and Lifestyle (AusDiab) study. Diabetologia 52: 415-424.
- BARR ELM ET Al. 2010. HOMA insulin sensitivity index and the risk of all-cause mortality and cardiovascular disease events in the general population: the Australian Diabetes, Obesity and Lifestyle Study (AusDiab) study. Diabetologia 53: 79-88.
- BASH LD, SELVIN E, STEFFES M, CORESH J and ASTOR BC. 2008. Poor glycemic control in diabetes and the risk of incident chronic kidney disease even in the absence of albuminuria and retinopathy: Atherosclerosis risk in communities (aric) study. Arch Intern Med 168: 2440-2447.
- BOIZEL R, BENHAMOU PY, LARDY B, LAPORTE F, FOULON T and HALIMI S. 2000. Ratio of triglycerides to HDL cholesterol is an indicator of LDL particle size in patients with type 2 diabetes and normal HDL cholesterol levels. Diabetes Care 23: 1679-1685.
- BONORA E ET AL. 2007. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in caucasian subjects from the general population: the Bruneck study. Diabetes Care 30: 318-324.
- BOWER RL and DL HAY. 2016. Amylin structure-function relationships and receptor pharmacology: implications for amylin mimetic drug development. Br J Pharmacol 173: 1883-1898.
- BOZZETTO L ET AL. 2016. Extra-Virgin Olive Oil Reduces Glycemic Response to a High-Glycemic Index Meal in Patients With Type 1 Diabetes: A Randomized Controlled Trial. Diabetes Care 39: 518-524.
- BRUDEVOLD R, HOLE T and HAMMERSTRØM J. 2008. Hyperferritinemia Is Associated with Insulin Resistance and Fatty Liver in Patients without Iron Overload. PLOS ONE 3: e3547.
- BUDOFF MJ ET AL. 2013. Progression of Coronary Calcium and Incident Coronary Heart Disease Events: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol 61: 1231-1239.
- BUTLER AE, JANSON J, BONNER-WEIR S, RITZEL R, RIZZA RA and BUTLER PC. 2003. β-Cell Deficit and Increased β-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes 52: 102-110.
- CABRERA DE LEÓN A ET AL. 2015. C-peptide as a risk factor of coronary artery disease in the general population. Diab Vasc Dis Res 12: 199-207.
- CAHILL GF. 2006. Fuel metabolism in starvation. Annu Rev Nutr 26: 1-22.
- CANELLA DS ET AL. 2014. Ultra-Processed Food Products and Obesity in Brazilian Households (2008-2009). PLOS ONE 9: e92752.
- CEFALU WT. 2016. "Prediabetes": Are There Problems With This Label? No, We Need Heightened Awareness of This Condition! Diabetes Care 39: 1472-1477.
- CHIA CW, ODETUNDE JO, KIM W, CARLSONO D, FERRUCCI L and EGAN JM. 2014. GIP Contributes to Islet Trihormonal Abnormalities in Type 2 Diabetes. J Clin Endocrinol Metab 99: 2477-2485.
- COLAGIURI S ET AL. 2011. Glycemic Thresholds for Diabetes-Specific Retinopathy Implications for diagnostic criteria for diabetes. Diabetes Care 34: 145-150.
- COOPER GJ, WILLIS AC, CLARK A, TURNER RC, SIM RB and REID KB. 1987. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A 84: 8628-8632.
- CORDAIN L, EADES MR and EADES MD. 2003. Hyperinsulinemic diseases of civilization: more than just Syndrome X. Comp Biochem Physiol A Mol Integr Physiol 136: 95-112.
- CRANE PK ET AL. 2013. Glucose Levels and Risk of Dementia. N Engl J Med 369: 540-548.
- CUNNANE SC and CRAWFORD MA. 2003. Survival of the fattest: fat babies were the key to evolution of the large human brain. Comp Biochem Physiol A Mol Integr Physiol 136: 17-26.
- DABELEA D ET AL. 2014. PRevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 311: 1778-1786.
- DCCT. 1993. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N Engl J Med 329: 977-986.
- DCCT-EDIC. 2000. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. N Engl J Med 342: 381-389.
- DECODE STUDY GROUP. 1999. Glucose tolerance and mortality: comparison of WHO and American Diabetic Association diagnostic criteria. The DECODE study group. European Diabetes Epidemiology Group. Diabetes Epidemiology: Collaborative analysis Of Diagnostic criteria in Europe. The Lancet 354: 617-621.
- DECODE STUDY GROUP, EUROPEAN DIABETES EPIDEMIOLOGY GROUP. 2003. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 26: 688-696.
- DEFRONZO RA. 1988. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 37: 667-687.
- DEFRONZO RA, TOBIN JD and ANDRES R. 1979. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237: E214-223.
- DESPRÉS JP ET AL. 1996. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med 334 :952-957.
- DEVECI E ET AL. 2009. Evaluation of insulin resistance in normoglycemic patients with coronary artery disease. Clin Cardiol 32: 32-36.
- DEWITT DE and IB HIRSCH. 2003. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: scientific review. JAMA 289:2254-2264.
- DOBBS R ET AL. 1975. Glucagon: role in the hyperglycemia of diabetes mellitus. Science 187: 544-547.
- DPP - DIABETES PREVENTION PROGRAM. 2002. Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin. Diabetes Prevention Program Research Group. N Engl J Med 346: 393-403.
- EBBELING CB, LEIDIG MM, FELDMAN HA, LOVESKY MM and LUDWIG DS. 2007. Effects of a low-glycemic load vs low-fat diet in obese young adults: a randomized trial. JAMA 297: 2092-2102.
- ESTRUCH R ET AL. 2016. Effect of a high-fat Mediterranean diet on bodyweight and waist circumference: a prespecified secondary outcomes analysis of the PREDIMED randomised controlled trial. Lancet Diabetes Endocrinol 4: 666-676.
-
ESTRUCH R ET AL. 2013. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet. research-article. <http://www.nejm.org/doi/full/10.1056/NEJMoa1200303> (22 August 2016).
» http://www.nejm.org/doi/full/10.1056/NEJMoa1200303 - FEDERATION. 2015. IDF Diabetes Atlas. P. In: International Diabetes Federation.
- FEINMAN RD ET AL. 2015. Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base. Nutr Burbank Los Angel Cty Calif 31: 1-13.
- FLOYD JC, FAJANS SS, PEK S and CHANCE RE. 1976. A newly recognized pancreatic polypeptide; plasma levels in health and disease. Recent Prog Horm Res 33: 519-570.
- FORGA LLENAS L, GOÑI IRIARTE MJ, CAMBRA CONTIN K, IBÁÑEZ BEROIZ B, CHUECA GUENDULAIN M and BERRADE ZUBIRI S. 2015. Incidence and temporal trends of childhood type 1 diabetes between 1975 and 2012 in Navarre (Spain). Gac Sanit SESPAS 29: 51-54.
- FOROUHI NG AND WAREHAM NJ. 2014. Epidemiology of diabetes. Medicine (Baltimore) 42: 698-702.
- GANNON MC and NUTTALL FQ. 2004. Effect of a high-protein, low-carbohydrate diet on blood glucose control in people with type 2 diabetes. Diabetes 53: 2375-2382.
- GARBER AJ ET AL. 2016. Consensus statement by the american association of clinical endocrinologists and american college of endocrinology on the comprehensive type 2 diabetes management algorithm - 2016 executive summary. Endocr Pract 22: 84-113.
- GARDNER CD ET AL. 2007. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA 297: 969-977.
- GARG MK, DUTTA MK and MAHALLE N. 2011. Study of beta-cell function (by HOMA model) in metabolic syndrome. Indian J Endocrinol Metab 15: S44-49.
- GASTALDELLI A, FERRANNINI E, MIYAZAKI Y, MATSUDA M, DEFRONZO RA and SAN ANTONIO METABOLISM STUDY. 2004. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia 47: 31-39.
- GODOY-MATOS AF. 2014. The role of glucagon on type 2 diabetes at a glance. Diabetol Metab Syndr 6: 91.
- GOTO A ET AL. 2016. High hemoglobin A1c levels within the non-diabetic range are associated with the risk of all cancers. Int J Cancer 138: 1741-1753.
- GRUNDY SM ET AL. 2005. Diagnosis and Management of the Metabolic Syndrome. Circulation 112: 2735-2752.
- GUERREIRO LH, DA SILVA D, SOLA-PENNA M, MIZURINI DM and LIMA LMTR. 2013. Amylin induces hypoglycemia in mice. An Acad Bras Cienc 85: 349-354.
- HALL KD ET AL. 2015. Calorie for Calorie, Dietary Fat Restriction Results in More Body Fat Loss than Carbohydrate Restriction in People with Obesity. Cell Metab 22: 427-436.
- HANLEY AJG, WILLIAMS K, STERN MP and HAFFNER SM. 2002. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care 25: 1177-1184.
- HARJUTSALO V, SJÖBERG L and TUOMILEHTO J. 2008. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. The Lancet 371: 1777-1782.
- HÄTÖNEN KA ET AL. 2006. Methodologic considerations in the measurement of glycemic index: glycemic response to rye bread, oatmeal porridge, and mashed potato. Am J Clin Nutr 84: 1055-1061.
- HAY DL, CHEN S, LUTZ TA, PARKES DG and ROTH JD. 2015. Amylin: Pharmacology, Physiology, and Clinical Potential. Pharmacol Rev 67: 564-600.
- HECHT HS. 2015. Coronary artery calcium scanning: past, present, and future. JACC Cardiovasc. Imaging 8: 579-596.
- HOLDEN SE ET AL. 2013. The incidence of type 2 diabetes in the United Kingdom from 1991 to 2010. Diabetes Obes Metab 15: 844-852.
- HUO X ET AL. 2016. Risk of non-fatal cardiovascular diseases in early-onset versus late-onset type 2 diabetes in China: a cross-sectional study. Lancet Diabetes Endocrinol 4: 115-124.
- IKEDA F ET AL. 2013. Haemoglobin A1c even within non-diabetic level is a predictor of cardiovascular disease in a general Japanese population: the Hisayama Study. Cardiovasc Diabetol 12: 164.
- INSTITUTE OF MEDICINE. 2005. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids (Macronutrients). P. in.: National Academies Press, Washington, D.C.
- INSTITUTE OF MEDICINE. 2006. Dietary Reference Intakes Research Synthesis: Workshop Summary. P. In: National Academies Press, Washington, D.C .
- JENKINS DJ ET AL. 1981. Glycemic index of foods: a physiological basis for carbohydrate exchange. Am J Clin Nutr 34: 362-366.
- JOHNSON JL, DUICK DS, CHUI MA and ALDASOUQI SA. 2010. Identifying prediabetes using fasting insulin levels. Endocr Pract 16: 47-52.
- JONES AG and HATTERSLEY AT. 2013. The clinical utility of C-peptide measurement in the care of patients with diabetes. Diabet Med 30: 803-817.
- KAHLEOVA H ET AL. 2014. Eating two larger meals a day (breakfast and lunch) is more effective than six smaller meals in a reduced-energy regimen for patients with type 2 diabetes: a randomised crossover study. Diabetologia 57: 1552-1560.
- KAHN SE ET AL. 1998. Reduced amylin release is a characteristic of impaired glucose tolerance and type 2 diabetes in Japanese Americans. Diabetes 47: 640-645.
- KARROWNI W ET AL. 2013. Insulin resistance is associated with significant clinical atherosclerosis in nondiabetic patients with acute myocardial infarction. Arterioscler Thromb Vasc Biol 33: 2245-2251.
- KATO M, NODA M, SUGA H, MATSUMOTO M and KANAZAWA Y. 2009. Fasting plasma glucose and incidence of diabetes --- implication for the threshold for impaired fasting glucose: results from the population-based Omiya MA cohort study. J Atheroscler Thromb 16: 857-861.
- KAUR B and HENRY J. 2014. Micronutrient status in type 2 diabetes: a review. Adv Food Nutr Res 71: 55-100.
- KHAW KT, WAREHAM N, BINGHAM S, LUBEN R, WELCH A and DAY N. 2004. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med 141: 413-420.
- KOHNER EM ET AL. 1998. United Kingdom Prospective Diabetes Study, 30: diabetic retinopathy at diagnosis of non-insulin-dependent diabetes mellitus and associated risk factors. Arch Ophthalmol 116(3): 297-303.
- KOPP W. 2003. High-insulinogenic nutrition--an etiologic factor for obesity and the metabolic syndrome? Metabolism 52: 840-844.
- KRAFT JR. 1975. Detection of Diabetes Mellitus In Situ (Occult Diabetes). Lab Med 6: 10-22.
- KRAFT JR. 2008. Diabetes Epidemic & You. P. In.: Trafford Publishing.
- KRIS-ETHERTON P, ECKEL RH, HOWARD BV, JEOR SS and BAZZARRE TL. 2001. Lyon Diet Heart Study. Circulation 103: 1823-1825.
- LAAKSO M. 1993. How good a marker is insulin level for insulin resistance? Am J Epidemiol 137: 959-965.
- LAMB MM, FREDERIKSEN B, SEIFERT JA, KROEHL M, REWERS M and NORRIS JM. 2015. Sugar intake is associated with progression from islet autoimmunity to type 1 diabetes: the Diabetes Autoimmunity Study in the Young. Diabetologia 58: 2027-2034.
- LARSSON H, BERGLUND G and AHRÉN B. 1995. Glucose modulation of insulin and glucagon secretion is altered in impaired glucose tolerance. J Clin Endocrinol Metab 80: 1778-1782.
- LEEMAN M, OSTMAN E and BJÖRCK I. 2005. Vinegar dressing and cold storage of potatoes lowers postprandial glycaemic and insulinaemic responses in healthy subjects. Eur J Clin Nutr 59: 1266-1271.
- LEROUX G ET AL. 2000. Influence of triglyceride concentration on the relationship between lipoprotein cholesterol and apolipoprotein B and A-I levels. Metabolism 49: 53-61.
- LI Y, LI Y, MENG L and ZHENG L. 2015. Association between Serum C-Peptide as a Risk Factor for Cardiovascular Disease and High-Density Lipoprotein Cholesterol Levels in Nondiabetic Individuals. PLoS ONE 10.
- LIM JS, MIETUS-SNYDER M, VALENTE A, SCHWARZ JM and LUSTIG RH. 2010. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol 7: 251-264.
- LIMA LMTR. 2017. Prediabetes definitions and clinical outcomes. Lancet Diabetes Endocrinol 5: 92-93.
- LINDAHL B, DINESEN B, ELIASSON M, RØDER M, HALLMANS G and STEGMAYR B. 2000. High Proinsulin Levels Precede First-Ever Stroke in a Nondiabetic Population. Stroke 31: 2936-2941.
- LONARDO A, BALLESTRI S, MARCHESINI G, ANGULO P and LORIA P. 2015. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig Liver Dis 47: 181-190.
- LOOKER HC ET AL. 2012. Diabetic retinopathy at diagnosis of type 2 diabetes in Scotland. Diabetologia 55: 2335-2342.
- LUCHSINGER JA TANG MX, SHEA S and MAYEUX R. 2004. Hyperinsulinemia and risk of Alzheimer disease. Neurology 63: 1187-1192.
- LUDVIK B, LELL B, HARTTER E, SCHNACK C and PRAGER R. 1991. Decrease of stimulated amylin release precedes impairment of insulin secretion in type II diabetes. Diabetes 40: 1615-1619.
- LUSTIG RH ET AL. 2016. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obes Silver Spring Md 24: 453-460.
- MARLISS EB, AOKI TT, UNGER RH, SOELDNER JS and CAHILL GF. 1970. Glucagon levels and metabolic effects in fasting man. J Clin Invest 49: 2256-2270.
- MATTHEWS DR, JP HOSKER, AS RUDENSKI, BA NAYLOR, DF TREACHER and RC TURNER. 1985. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412-419.
- MAZUR A. 2011. Why were "starvation diets" promoted for diabetes in the pre-insulin period? Nutr J 10: 23.
- MCCLAIN AD, OTTEN JJ, HEKLER EB and GARDNER CD. 2013. Adherence to a low-fat vs. low-carbohydrate diet differs by insulin resistance status. Diabetes Obes Metab 15: 87-90.
- MECHANICK JI. 2015. Global Dimensions of Diabetes: Information and Synthesis. Ann Glob Health 81: 733-734.
- MENKE A, CASAGRANDE S, GEISS L and COWIE CC. 2015. Prevalence of and trends in diabetes among adults in the United States, 1988-2012. JAMA 314: 1021-1029.
- MIAO X, SUN W, FU Y, MIAO L and CAI L. 2013. Zinc homeostasis in the metabolic syndrome and diabetes. Front Med 7: 31-52.
- MOZAFFARIAN D and JHY WU. 2012. (n-3) fatty acids and cardiovascular health: are effects of EPA and DHA shared or complementary? J Nutr 142: 614S-625S.
- MUTI P ET AL. 2002. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomarkers Prev 11: 1361-1368.
- NAGI DK, PETTITT DJ, BENNETT PH, KLEIN R and KNOWLER WC. 1997. Diabetic retinopathy assessed by fundus photography in Pima Indians with impaired glucose tolerance and NIDDM. Diabet Med J Br Diabet Assoc 14: 449-456.
- NATHAN DM ET AL. 2005. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643-2653.
- NCD-RISC. 2016. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4·4 million participants. NCD Risk Factor Collaboration (NCD-RisC). The Lancet 387: 1513-1530.
- NICHOLS GA, HILLIER TA and BROWN JB. 2007. Progression from newly acquired impaired fasting glusose to type 2 diabetes. Diabetes Care 30: 228-233.
- NICHOLS GA, HILLIER TA and BROWN JB. 2008. Normal fasting plasma glucose and risk of type 2 diabetes diagnosis. Am J Med 121: 519-524.
- OHN JH ET AL. 2016. 10-year trajectory of β-cell function and insulin sensitivity in the development of type 2 diabetes: a community-based prospective cohort study. Lancet Diabetes Endocrinol 4: 27-34.
- ONIKI K ET AL. 2016. The longitudinal effect of the aldehyde dehydrogenase 2*2 allele on the risk for nonalcoholic fatty liver disease. Nutr Diabetes 6: e210.
- OSTMAN E, GRANFELDT Y, PERSSON L and BJÖRCK I. 2005. Vinegar supplementation lowers glucose and insulin responses and increases satiety after a bread meal in healthy subjects. Eur J Clin Nutr 59: 983-988.
- PAIS R ET AL. 2016. Fatty liver is an independent predictor of early carotid atherosclerosis. J Hepatol 65(1): 95-102.
- PATTERSON CC, DAHLQUIST GG, GYÜRÜS E, GREEN A and SOLTÉSZ G. 2009. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. The Lancet 373: 2027-2033.
- PETERSEN KF ET AL. 2007. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A 104: 12587-12594.
- POLONSKY KS, GIVEN BD and VAN CAUTER E. 1988. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest 81: 442-448.
-
POURFARZIB R ET AL. 2014. Relationship between plasma apolipoprotein B concentrations and LDL particle number. Research Reports in Clinical Cardiology. <https://www.dovepress.com/relationship-between-plasma-apolipoprotein-b-concentrations-and-ldl-pa-peer-reviewed-article-RRCC> (14 June 2016).
» https://www.dovepress.com/relationship-between-plasma-apolipoprotein-b-concentrations-and-ldl-pa-peer-reviewed-article-RRCC - REAVEN GM, FARQUHAR JW and NAKANISHI RH. 1969. Steady State Plasma Insulin Response to Continuous Glucose Infusion in Normal and Diabetic Subjects. Diabetes 18: 273-279.
- REAVEN GM, LERNER RL, STERN MP and FARQUHAR JW. 1967. Role of insulin in endogenous hypertriglyceridemia. J Clin Invest 46: 1756-1767.
- ROBERTSON MD, HENDERSON RA, VIST GE and RUMSEY RDE. 2002. Extended effects of evening meal carbohydrate-to-fat ratio on fasting and postprandial substrate metabolism. Am J Clin Nutr 75: 505-510.
- RYDÉN L ET AL. 2007. Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: executive summary. Eur Heart J 28: 88-136.
- RYDÉN L, VIVECA G, SCHNELL O and JAAKKO T. 2016. Oral glucose tolerance testing and cardiovascular disease. Lancet Diabetes Endocrinol 4: 732-733.
- SALAS-SALVADÓ J ET AL. 2011. Reduction in the Incidence of Type 2 Diabetes With the Mediterranean Diet. Diabetes Care 34: 14-19.
- SANYAL D, MUKHERJEE P, RAYCHAUDHURI M, GHOSH S, MUKHERJEE S and CHOWDHURY S. 2015. Profile of liver enzymes in non-alcoholic fatty liver disease in patients with impaired glucose tolerance and newly detected untreated type 2 diabetes. Indian J Endocrinol Metab 19: 597-601.
- SCHMIDT MI ET AL. 2011. Chronic non-communicable diseases in Brazil: burden and current challenges. The Lancet 377: 1949-1961.
- SCHWARTZ SS, EPSTEIN S, CORKEY BE, GRANT SFA, GAVIN JR and AGUILAR RB. 2016. The Time Is Right for a New Classification System for Diabetes: Rationale and Implications of the β-Cell-Centric Classification Schema. Diabetes Care 39: 179-186.
- SCHWARZ JM ET AL. 2015. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J Clin Endocrinol Metab 100: 2434-2442.
- SELVIN E, CORESH J, GOLDEN SH, BRANCATI FL, FOLSOM AR and STEFFES MW. 2005a. Glycemic Control and Coronary Heart Disease Risk in Persons With and Without Diabetes: The Atherosclerosis Risk in Communities Study. Arch Intern Med 165: 1910.
- SELVIN E, CORESH J, SHAHAR E, ZHANG L, STEFFES M and SHARRETT AR. 2005b. Glycaemia (haemoglobin A1c) and incident ischaemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Lancet Neurol 4: 821-826.
- SELVIN E ET AL. 2010. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 362: 800-811.
- SHALAUROVA I, CONNELLY MA, GARVEY WT and OTVOS JD. 2014. Lipoprotein insulin resistance index: a lipoprotein particle-derived measure of insulin resistance. Metab Syndr Relat Disord 12: 422-429.
- SHAW JE ET AL. 2000. Impaired fasting glucose: how low should it go? Diabetes Care 23: 34-39.
- SIERI S ET AL. 2012. Prospective study on the role of glucose metabolism in breast cancer occurrence. Int J Cancer 130: 921-929.
- SIMMONS D and HLAING T. 2014. Interpretation of HbA1c: association with mean cell volume and haemoglobin concentration. Diabet Med 31: 1387-1392.
- SISNANDE T ET AL. 2015. Monoconjugation of Human Amylin with Methylpolyethyleneglycol. PloS One 10: e0138803.
- SMITS MM, IOANNOU GN, BOYKO EJ and UTZSCHNEIDER KM. 2013. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: results of a US national survey in three ethnic groups. J Gastroenterol Hepatol 28: 664-670.
- SONG SH. 2016. Early-onset type 2 diabetes: high lifetime risk for cardiovascular disease. Lancet Diabetes Endocrinol 4: 87-88.
- STEVEN S ET AL. 2016. Very-Low-Calorie Diet and 6 Months of Weight Stability in Type 2 Diabetes: Pathophysiologic Changes in Responders and Nonresponders. Diabetes Care 39(5): 808-815.
- STRATTON IM ET AL. 2000. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321: 405-412.
- THOMASETH K ET AL. 1997. Amylin release during oral glucose tolerance test. Diabet Med J Br Diabet Assoc 14(Suppl 2): S29-34.
- TIROSH A ET AL. 2005. Normal fasting plasma glucose levels and type 2 diabetes in young men. N Engl J Med 353: 1454-1462.
- TOSCANO CM ET AL. 2015. Cost-effectiveness of a national population-based screening program for type 2 diabetes: the Brazil experience. Diabetol Metab Syndr 7: 95.
- TÓTH C and CLEMENS Z. 2015. A child with type 1 diabetes mellitus (T1DM) successfully treated with the Paleolithic ketogenic diet: A 19-month insulin-freedom. Int J Case Rep Images 6: 752.
- UKPDS. 1998a. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet Lond Engl 352: 854-865.
- UKPDS. 1998b. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet Lond Engl 352: 837-853.
- UNGER RH AND ORCI L. 1975. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet Lond Engl 1:14-16.
- UNWIN DJ, CUTHBERTSON DJ, FEINMAN RD and SPRUNG VS. 2015. A pilot study to explore the role of a low-carbohydrate intervention to improve GGT levels and HbA1c. Diabesity in Practice 4: 102-108.
-
USDA. 2014. Nutrient Data : SR27. USDA - United States Department of Agriculture, ARS - Agricultural Research Service. <https://www.ars.usda.gov/Services/docs.htm?docid=24912> (21 April 2016).
» https://www.ars.usda.gov/Services/docs.htm?docid=24912 - VALENTI V ET AL. 2015. A 15-Year Warranty Period for Asymptomatic Individuals Without Coronary Artery Calcium: A Prospective Follow-Up of 9,715 Individuals. JACC Cardiovasc Imaging 8: 900-909.
- VALENTI V ET AL. 2016. Absence of Coronary Artery Calcium Identifies Asymptomatic Diabetic Individuals at Low Near-Term But Not Long-Term Risk of Mortality A 15-Year Follow-Up Study of 9715 Patients. Circ Cardiovasc Imaging 9: e003528.
- VAN'T RIET E ET AL. 2012. HbA1c is an independent predictor of non-fatal cardiovascular disease in a Caucasian population without diabetes: a 10-year follow-up of the Hoorn Study. Eur J Prev Cardiol 19: 23-31.
- WALLACE TM, LEVY JC and MATTHEWS DR. 2004. Use and Abuse of HOMA Modeling. Diabetes Care 27: 1487-1495.
- WEISS R, BREMER AA and LUSTIG RH. 2013. What is metabolic syndrome, and why are children getting it? Ann N Y Acad Sci 1281: 123-140.
- WESTERMARK P, WERNSTEDT C, WILANDER E, HAYDEN DW, O'BRIEN TD and JOHNSON KH. 1987. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A 84: 3881-3885.
-
WHO. 2015. Health topics - Diabetes. World Health Organizaiton (WHO). WHO. <http://www.who.int/topics/diabetes_mellitus/en/> (24 December 2015a).
» http://www.who.int/topics/diabetes_mellitus/en/ -
WHO. 2016. Use of glycated haemoglobin (HbA1c) in the diagnosis of diabetes mellitus. World Health Organization (WHO). WHO. <http://www.who.int/diabetes/publications/diagnosis_diabetes2011/en/> (14 February 2016b).
» http://www.who.int/diabetes/publications/diagnosis_diabetes2011/en/ - WIENER K. 1995. Fasting plasma glucose as a diagnostic indicator of diabetes mellitus. Clin Chim Acta Int J Clin Chem 238: 199-208.
- WONG ND ET AL. 2003. The metabolic syndrome, diabetes, and subclinical atherosclerosis assessed by coronary calcium. J Am Coll Cardiol 41: 1547-1553.
- WONG ND ET AL. 2012. Metabolic Syndrome, Diabetes, and Incidence and Progression of Coronary Calcium: The Multiethnic Study of Atherosclerosis Study. JACC Cardiovasc Imaging 5: 358-366.
- WU A. 2006. Tietz Clinical Guide to Laboratory Tests Fourth Edition. P. In.: Saunders, St. Louis, Mo.
- YALOW RS and SA BERSON. 1960. Immunoassay of endogenous plasma insulin in man. J. Clin. Invest. 39: 1157-1175.
- YALOW RS , GLICK SM, ROTH J and BERSON SA. 1965. Plasma insulin and growth hormone levels in obesity and diabetes. Ann N Y Acad Sci 131: 357-373.
- YOUNG A. 2005. Inhibition of glucagon secretion. Adv Pharmacol San Diego Calif 52: 151-171.
- YUDKIN JS. 2016. "Prediabetes": Are There Problems With This Label? Yes, the Label Creates Further Problems! Diabetes Care 39: 1468-1471.
- YUDKIN JS and MONTORI VM. 2014. The epidemic of pre-diabetes: the medicine and the politics. BMJ 349: g4485.
- ZEEVI D ET AL. 2015. Personalized Nutrition by Prediction of Glycemic Responses. Cell 163: 1079-1094.
- ZHANG P, ENGELGAU MM, VALDEZ R, CADWELL B, BENJAMIN SM and NARAYAN KMV. 2005. Efficient cutoff points for three screening tests for detecting undiagnosed diabetes and pre-diabetes: an economic analysis. Diabetes Care 28: 1321-1325.
- ZILKER TR ET AL. 1988. Pharmacokinetics of biosynthetic human proinsulin following intravenous and subcutaneous administration in metabolically healthy volunteers. Horm Metab Res Suppl Ser 18: 37-43.
-
2
"Diabetes is a group of metabolic diseases characterized by hyperglycaemia resulting from defects in insulin secretion, insulin action, or both." American Diabetes Association (Association 2014)
-
3
"The definition and diagnostic classification of diabetes and its pre-states should be based on the level of the subsequent risk of cardiovascular complications". Guidelines on diabetes, pre-diabetes, and cardiovascular diseases: executive summary. European Society of Cardiology (ESC) (Rydén et al. 2007)
-
4
"Insulin resistance is prerequisite for the development of type 2 diabetes and becomes manifest long before hyperglycaemia is evident.". "Insulin-resistant individuals develop type 2 diabetes only if β-cell failure ensues. Thus, measures of β-cell function might be expected to be a key predictor for future type 2 diabetes.". (Abdul-Ghani and DeFronzo 2009)
-
5
"Those with cardiovascular disease not identified with diabetes are simply undiagnosed". (Kraft 2008)
-
6
"Islet dysfunction is critical for development of IGT and type 2 diabetes and (...) these pathophysiological events already start when subjects are normal glucose tolerant". (Ahrén, 2009)
-
8
"First line "treatment" for pre-diabetes by whatever definition is lifestyle advice. And because the risk factors overlap with those of other non-communicable diseases, the question is why focus attention on a specific group of people with a diagnosis of pre-diabetes while ignoring the remainder of the healthy population who would benefit from the same advice.". Yudkin and Montori 2014
-
9
"From a clinical perspective, the most practical way of assessing insulin resistance would seem to be the measurement of insulin concentration in plasma". "Plasma insulin levels whether measured in the fasting state or after a glucose load are a powerful predictor for the risk of type 2 diabetes". Consensus Development Conference on Insulin Resistance American Diabetes Association (Association 1998)