Abstracts
PURPOSE: The aim of this work was to investigate the hypothesis that catechol inhibits FADH2-linked basal respiration in mitochondria isolated from rat liver homogenates. Moreover, catechol ability to induce peroxidation of biomolecules in liver nuclear fractions was also studied. METHODS: Rat liver homogenates were incubated with 1mM 1,2-dihydroxybenzene (catechol) at pH 7.4 for up to 30 minutes. After that, mitochondrial fractions were isolated by differential centrifugation. Basal oxygen uptake was measured using a Clark-type electrode after the addition of 10 mM sodium succinate. Nuclear fractions were incubated in the presence of 1 mM catechol for 17 hours at room temperature and the peroxidation of biomolecules was investigated by the reaction with thiobarbituric acid, which was determined spectrophotometrically at 535 nm. RESULTS: Catechol induced a time-dependent partial inhibition of FADH2-linked basal mitochondrial respiration, however this substance was unable to induce a direct peroxidation of biomolecules in hepatic nuclear fractions. CONCLUSION: Catechol produced an inhibition of basal respiration associated to FADH2 in isolated liver mitochondria that could lead to cytotoxicity, ROS generation and cell death.
Catechol; Mitochondrial respiration; Peroxidation
OBJETIVO: Testar a hipótese do catecol inibir a respiração basal associada ao FADH2 em frações mitocondriais hepáticas de rato. Além disso, estudou-se também a capacidade do catecol de induzir peroxidação de biomoléculas nas frações nucleares. MÉTODOS: Os homogeneizados de fígado de ratos foram incubados com catecol a 1 mM em pH fisiológico. Depois disso, as frações mitocondriais foram isoladas por centrifugação diferencial. O consumo basal de oxigênio foi medido com um eletrodo do tipo Clark após injeção de succinato a 10 mM. Frações nucleares foram incubadas com catecol por 17 horas à temperatura ambiente e a peroxidação de biomoléculas foi investigada pela reação com o ácido tiobarbitúrico e mensurada espectrofotometricamente. RESULTADOS: O catecol induziu uma inibição parcial da respiração basal mitocondrial associada ao FADH2 de forma dependente do tempo, contudo essa substância não induziu peroxidação direta das biomoléculas presentes nas frações nucleares hepáticas. CONCLUSÃO: O catecol produz inibição da respiração basal associada ao FADH2 em mitocôndrias isoladas de fígado, o que pode levar à toxicidade, produção de espécies reativas e morte celular.
Catecol; Respiração mitocondrial; Peroxidação
Catechol inhibits FADH2-linked respiration in rat liver mitochondrial fraction1 1 Laboratory of Neurochemistry and Cell Biology (LabNq), Health Sciences Institute (ICS), Federal University of Bahia (UFBA), Salvador - BA - Brazil.
Catecol inibe FADH2 ligado à respiração na fração mitochondrial do fígado do rato
George Emílio Sampaio BarretoI; Gleide Souza dos SantosII; Eryvaldo Sócrates Tabosa EgitoIII; Ramon dos Santos El-BacháIV
IPhysiotherapist, specialist in Respiratory Care, M. Sc. student of the Graduation Program in Health Sciences, Laboratory of Dispersed Systems (LASID), Department of Pharmacy, Federal University of Rio Grande do Norte (UFRN), Natal RN - Brazil
IIUndergraduate student of Nursing, UFBA, Fellowship of FAPESB, LabNq, ICS,UFBA, Salvador - BA - Brazil
IIIPh. D., Head of Pharmacy Course, Head of LASID, Department of Pharmacy-UFRN, Professor of Postgraduate Program of Health Sciences-UFRN, Natal - RN - Brazil
IVPh. D., Head of LabNq, Department of Biochemistry and Biophysics, ICS, UFBA, Salvador BA - Brazil
Correspondence Correspondence to Ramon dos santos El-Bachá Mailing address: ramon@ufba.br.
ABSTRACT
PURPOSE: The aim of this work was to investigate the hypothesis that catechol inhibits FADH2-linked basal respiration in mitochondria isolated from rat liver homogenates. Moreover, catechol ability to induce peroxidation of biomolecules in liver nuclear fractions was also studied.
METHODS: Rat liver homogenates were incubated with 1mM 1,2-dihydroxybenzene (catechol) at pH 7.4 for up to 30 minutes. After that, mitochondrial fractions were isolated by differential centrifugation. Basal oxygen uptake was measured using a Clark-type electrode after the addition of 10 mM sodium succinate. Nuclear fractions were incubated in the presence of 1 mM catechol for 17 hours at room temperature and the peroxidation of biomolecules was investigated by the reaction with thiobarbituric acid, which was determined spectrophotometrically at 535 nm.
RESULTS: Catechol induced a time-dependent partial inhibition of FADH2-linked basal mitochondrial respiration, however this substance was unable to induce a direct peroxidation of biomolecules in hepatic nuclear fractions.
CONCLUSION: Catechol produced an inhibition of basal respiration associated to FADH2 in isolated liver mitochondria that could lead to cytotoxicity, ROS generation and cell death.
Keywords: Catechol. Mitochondrial respiration. Peroxidation.
RESUMO
OBJETIVO: Testar a hipótese do catecol inibir a respiração basal associada ao FADH2 em frações mitocondriais hepáticas de rato. Além disso, estudou-se também a capacidade do catecol de induzir peroxidação de biomoléculas nas frações nucleares.
MÉTODOS: Os homogeneizados de fígado de ratos foram incubados com catecol a 1 mM em pH fisiológico. Depois disso, as frações mitocondriais foram isoladas por centrifugação diferencial. O consumo basal de oxigênio foi medido com um eletrodo do tipo Clark após injeção de succinato a 10 mM. Frações nucleares foram incubadas com catecol por 17 horas à temperatura ambiente e a peroxidação de biomoléculas foi investigada pela reação com o ácido tiobarbitúrico e mensurada espectrofotometricamente.
RESULTADOS: O catecol induziu uma inibição parcial da respiração basal mitocondrial associada ao FADH2 de forma dependente do tempo, contudo essa substância não induziu peroxidação direta das biomoléculas presentes nas frações nucleares hepáticas.
CONCLUSÃO: O catecol produz inibição da respiração basal associada ao FADH2 em mitocôndrias isoladas de fígado, o que pode levar à toxicidade, produção de espécies reativas e morte celular.
Descritores: Catecol. Respiração mitocondrial. Peroxidação.
Introduction
Benzene is an ubiquitous environmental chemical that is used as a precursor in the synthesis of numerous products including drugs, dyes, insecticides, and plastics. This compound is also found in unleaded gasoline, cigarette smoke and industrial emissions 1. Based on epidemiological studies, human exposure to benzene causes bone marrow depression, acute myelogenous leukemia, acute lymphocytic leukemia, myelotoxicity, non-Hodgkin´s lymphoma, lung cancer and nasopharyngeal cancer 1,2. The liver is the primary site of benzene metabolism and the major metabolic enzyme system is cytochrome P450 (primarily CYP2E1). This involves the formation of a series of reactive metabolites like phenol, hydroquinone and 1,2-dihydroxybenzene (catechol) 2.
Catechols are constituted by a large group of compounds from natural or synthetic origin, all them containing the common 1,2-dihydroxybenzene ring. They are used in a variety of applications, such as a reagent for photography, dyes, rubber and plastic production, and in the pharmaceutical industry 3. Catechols are intermediary products from the degradation of aromatic compounds. In humans and other mammals, catechols can occur as metabolites in the degradation of benzene and estrogenss4.
Catechols readily undergo oxidation to form semiquinone radicals and quinones, which are more reactive than catechols. The mechanisms most frequently cited to explain the toxicity of catechols are: (i) the generation of reactive oxygen species (ROS) by redox reactions; (ii) DNA damage in the form of oxidative damage or DNA arylation; (iii) protein damage by sulfhydryl arylation or oxidation; and (iv) interference with electron transport in energy transducing membranes 5. Moreover, catechol releases iron from ferritin inducing lipid peroxidation in brain homogenates 6. When catechol is oxidized enzimatically or in the presence of oxygen and heavy metals, one electron is transferred to the molecular oxygen, and consequently superoxide (O2-) is formed. In the presence of heavy metals (e.g. copper, iron), superoxide is further reduced to hydrogen peroxide (H2O2) and hydroxyl radicals (·OH). These ROS can be harmful to cells and organisms if they are not eliminated 7. Catechol can act as pro-oxidant damaging macromolecules such as DNA and proteins, and destroying membrane functioning due to their redox cycling activity. Our laboratory recently reported that catechol-induced cytotoxicity towards glioblastoma cells in vitro is due to the production of superoxide and reactive quinones 8.
Although the exact mechanism for benzene-induced toxicity is unknown, it seems that catechol plays an important role regarding the effects of the exposure to this molecule. The lethal human dose of catechol is 454 - 4540 mmol/kg or one teaspoon given once for a 70 kg person. Therefore, in the present study we investigate the toxic effect of 1 mM catechol towards liver subcellular fractions since this organ is responsible for xenobiotic metabolism. This concentration is in the range of the catechol level found accumulated in people exposed to action of this compound (90 - 9000 mM) 9. The hypothesis that catechol inhibits mitochondrial state 2 FADH2-linked respiration that could be a mechanism of toxicity was tested. Furthermore, the ability of catechol to induce an oxidative stress and a direct peroxidation of biomolecules due to its autoxidation was assessed.
Methods
Animals
Adult Wistar rats weighing 250-350 g were obtained from the Department of Physiology of the Health Sciences Institute of the Federal University of Bahia (Salvador, BA, Brazil). All experimental protocols were conducted according to regulations suggested by the Federal University of Bahia Ethical Committee.
Mitochondrial isolation
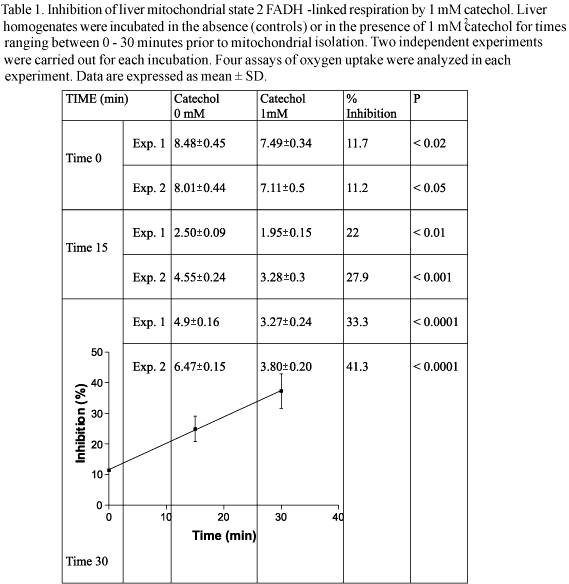
Mitochondria were isolated from liver of adult rats by differential centrifugation. Liver was homogenized in 0.5 M mannitol, 1 M sucrose, 0.2 M ethylene glycol-bis[b-aminoethyl ether]-N,N,N',N'-tetraacetic acid (EGTA), 0.05% (w/v) bovine serum albumin (BSA) and 1 M Tris buffer (pH 7.4) using a teflon pestle tissue homogenizer. Homogenates were incubated in the absence (controls) or in the presence of 1 mM catechol for 0, 15 or 30 minutes at 37 °C before centrifugation. Two independent experiments were done for each incubation. Whole cells, nuclei, cytoskeletons and plasma membranes were removed by centrifugation at 550 g for 10 minutes at 4 °C, followed by centrifugation of the supernatant at 7,100 g for 10 minutes at the same temperature. The mitochondrial pellet was resuspended in the same buffer and washed twice at 6,400 g for 10 minutes at 4 °C. Finally, mitochondria were resuspended in 1 ml of the same isolation buffer. Protein determinations were performed according to Lowry 10.
Oxygen electrode measurements
Oxygen consumption was carried out at 37 °C in a closed chamber containing a Clark type oxygen electrode connected to a YSI model 53 monitor (Yellow Springs Instrument Co. Inc., OH, USA). Isolated mitochondria were suspended in 3 ml of 10 mM KCl, 0.2 mM ethylenediamine tetraacetic acid (EDTA), 0.25 M mannitol, 0.025% (w/v) BSA and 10 mM Tris, 5 mM phosphate buffer (pH 7.4) at a final concentration of 0.4 mg protein/ml. Sodium succinate was added to a final concentration of 10 mM in order to induce mitochondrial basal respiration (state 2). Oxygen uptake in resting conditions was monitored for 12 minutes. Four assays were analyzed for each mitochondrial fraction.
Isolation of nuclear fraction
Nuclear fraction was isolated from liver of adult rats. To isolate nuclei, liver was weighed and suspended (0.3g/ml) in 0.25 M sucrose, 10 mM EDTA and 50 mM phosphate buffer (pH 7.4). The tissue was homogenized on ice using a teflon pestle tissue homogenizer. Cells were lysed with 8 up-and-down pestle strokes. Tissue homogenate was centrifuged at 400g for 10 minutes at 4 °C and the supernatant was stored at 4 °C. The pellet was resuspended in the same buffer and centrifuged again in the same conditions. Supernatants were combined and centrifuged at 1,500g for 10 minutes at 4 °C. Nuclei were resuspended in 1 mM EDTA, 20% glycerol and 100 mM phosphate buffer (pH 7.4). Protein concentrations were determined as described for mitochondrial isolation.
Peroxidation assay
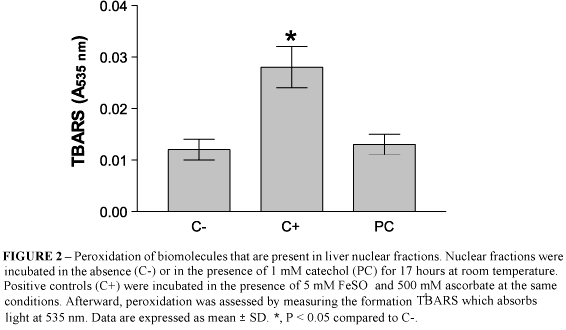
Nuclear fractions (1 mg/ml) were incubated with 1 mM catechol in 1 mM HCl and 50 mM phosphate buffer (pH 7.4) at room temperature for 17 hours. Negative controls were incubated in the absence of catechol and positive controls were incubated in the presence of 5 mM FeSO4 and 500 mM ascorbate. Peroxidation was assessed by measuring the formation of thiobarbituric acid-reactive substances (TBARS) as described previously 11. A great variety of oxidized substances form pink TBA complexes, such as malonaldehyde, oxidized sugars and amino acids 12, and all these compounds can be found in nuclear fractions submitted to an oxidative stress. After addition of 2 ml of 0.67% (w/v) TBA, 15% (w/v) trichloroacetic acid in 0.25 M HCl, samples were boiled in capped tubes for 10 minutes and cooled on fresh water. The reaction mixture was centrifuged at 1,500 g for 10 minutes. The optical density was measured against a blank without TBA in a Micronal B 382 spectrophotometer at 535 nm .
Statistics
Samples and controls were compared by the Student's t test. Significant differences were considered for P < 0.05. Pearson's test was used to correlate catechol-induced inhibition of mitochondrial respiration with time.
Results
Catechol inhibited mitochondrial state 2 FADH2-linked respiration when liver homogenates were incubated in the presence of this compound (Table 1). The inhibition (11.5%) was evident even when mitochondrial fractions were isolated just after the addition of 1 mM catechol (Time 0). The inhibition increased when homogenates were incubated with this molecule for 15 minutes (25 %) or 30 minutes (37.3%) before mitochondrial isolation. Moreover, the inhibition linearly correlates with time (Pearson's correlation, 0.96458; P < 0.002; Figure 1).
Table 1. Inhibition of liver mitochondrial state 2 FADH2-linked respiration by 1 mM catechol. Liver homogenates were incubated in the absence (controls) or in the presence of 1 mM catechol for times ranging between 0 - 30 minutes prior to mitochondrial isolation. Two independent experiments were carried out for each incubation. Four assays of oxygen uptake were analyzed in each experiment. Data are expressed as mean ± SD.
In order to investigate if catechol could induce a direct peroxidation of biomolecules, nuclear fractions were incubated with this substance at 1 mM for 17 hours at room temperature and TBARS were measured after this. Catechol did not induce peroxidation of biomolecules present in nuclear fractions (Figure 2). On the other hand, a marked peroxidation was observed in samples incubated in the presence of ascorbate and FeSO4 (positive controls).
Discussion
Data from the present study demonstrate that catechol inhibits liver mitochondrial state 2 FADH2-linked respiration. These data suggest the inhibition of mitochondrial respiration by catechol as a potential mechanism of its cytotoxicity. Other laboratories have shown that endogenous catechols and exogenous molecules bearing a catechol moiety are also inhibitors of mitochondrial respiration. Endogenous cysteinylcatechols are potent inhibitors of mitochondrial complex I activity in vitro 13. Dopamine and its metabolite 3,4-dihydroxyphenylacetic acid can inhibit brain mitochondrial state 3 NADH-linked respiration 14. Flavonoids with a catechol on their rings were inhibitors of state 2 FADH2- and NADH-linked respiration 15. A previous study about catechol-O-methyltransferase inhibitors demonstrated the effects of 3,4-dihydroxy-4'-methyl-5-nitrobenzophenone (tolcapone) in decreasing respiratory control ratio in mitochondrial preparations at low micromolar concentrations 16. Tolcapone has been associated with hepatotoxicity. Furthermore, tolcapone reduced ATP synthesis in human neuroblastoma SH-SY5Y cells 17.
Excessive generation of reactive oxygen species (ROS) is presumed to be a significant factor in tissue injury observed in many disease states. There are numerous sites of ROS production within cells including cytochrome P450 enzymes, xanthine oxidase, and the mitochondrial electron transport chain. It has been shown that mitochondria generate superoxide and hydrogen peroxide during state 4 respiration at levels which constitute about 2% of total mitochondrial oxygen uptake. Mitochondrial production of ROS is due to reduction of molecular oxygen by an electron that "leaks" from the unstable ubiquinone semiquinone anion that is formed during redox cycling of ubiquinone present in mitochondrial complex III 18. Furthermore, mitochondrial ROS may inhibit one or more of the components of the respiratory chain, further accelerating the rate of superoxide formation. The molecular targets of mitochondrial-derived ROS have not been clearly established, although lipid peroxidation, ion channel modification and DNA damage have all been demonstrated in models where the effects of exogenous oxidizing agents have been studied 19.
Catechol leads to the formation of ROS and reactive quinones during its autoxidation. Quinones may contribute to increase H2O2 production in mitochondria, as reported in subcellular fractions treated with estrone 3,4-quinone and NADPH 20. Oxidative stress increases the opening of mitochondrial permeability transition pore (mPTP) and depolarisation 21, suggesting that this mechanism may be important in cell death induced by catechol.
In our experiments catechol did not induce a direct peroxidation of biomolecules in liver nuclear fractions. Indeed, other authors showed that catechols such as catechol estrogens, catecholamines, catechins and caffeic acid strongly inhibit lipid peroxidation 22. However, catechol releases iron from ferritin inducing lipid peroxidation in brain homogenates 6. Although the pro-oxidant or antioxidant properties of catechols rests a controversial subject, our data show that the inhibition of mitochondrial respiration induced by catechol is more important than the peroxidation of biomolecules in liver subcellular fractions.
Benzene is metabolized in the liver to catechol by hepatic cytochrome P450 enzymes, particularly CYP2E1 23-24. Exposure to benzene has been associated with perisinusoidal fibrosis, histological evidence of cholestasis, and non-alcoholic steatohepatitis 25. Overall, the results from this study permit us to propose that benzene toxicity is related to formation of catechol which kills hepatocytes by inhibiting FADH2-linked mitochondrial respiration.
Conclusion
The catechol level found accumulated in people exposed to action of this compound is in the range of 90 - 9000 mM. This study showed that catechol at 1 mM inhibits state 2 FADH2-linked mitochondrial respiration. However, this molecule did not induce peroxidation in nuclear fractions. These data suggest that catechols may exert hepatotoxicity by inhibiting mitochondrial respiration. This mechanism may be related to benzene-induced non-alcoholic steatohepatitis, since catechol is one of its main metabolites.
Acknowledgments
This work was supported by grants from FAPESB (317/2003), CNPq (472341/2001), and BNB (2002-1-502). We are grateful to Mr. C. A. R. Silva for his technical assistance.
This work was supported by UFBA, FAPESB, CNPq and BNB/FUNDECI
- 1.Golding BT, Watson WP. Possible mechanisms of carcinogenesis after exposure to benzene. IARC Scientific Pub 1999; 150: 75-88.
- 2.Snyder R. Benzene and Leukemia. Crit Rev Toxicol 2002; 32: 155-210.
- 3.Millingan PW, Hãggblom MM. Biodegradation of resorcinol and catechol by denitrifying enrichment cultures. Environ Toxicol Chem 1998; 17: 1456-71.
- 4.Bolton JL, Pisha E, Zhang F, Qiu S. Role of quinoids in estrogen carcinogenesis. Chem Res Toxicol 1998; 11: 1113-27.
- 5.Schweigert N, Hunziker R, Escher BI, Eggen RIL. The acute toxicity of (chloro-) catechol and (chloro-) catechol-cooper combination in Escherichia coli corresponds to the membrane toxicity of these compounds. Environ Toxicol Chem 2001; 20: 239-47.
- 6.Agrawal R, Sharma PK, Rao GS. Release of iron from feritin by metabolites of benzene and superoxide radical generating agents. Toxicol 2001; 168: 223-30.
- 7.Schweigert N, Acero JL, Von Gunten U, Canonica S, Zehnder AJB, Eggen RIL. DNA degradation by the mixture of cooper and catechol is caused by DNA-cooper-oxo complexes, probably DNA-cu (I) OOH. Environ Mol Mutagen 2000; 36: 5-12.
- 8.Pereira MRG, de Oliveira ES, de Villar FAGA, Grangeiro MS, Fonseca J, Silva AR, Costa MFD, Costa SL, El-Bachá RS. Cytotoxicity of catechol towards human glioblastoma cells via superoxide and reactive quinones generation. J Bras Patol Med Lab 2004; 40: 281-6.
- 9.Bukowska B, Kowalska S. Phenol and catechol induce prehemolytic and hemolytic changes in human erythrocytes. Toxicol Lett 2004; 152: 73-84.
- 10.Lowry O, Rosenbrough NJ, Farr A, Randan RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951; 193: 265-75.
- 11.Slater TF, Overview of methods used for detecting lipid peroxidation., In: Packer L. Oxygen radicals in biological systems. Orlando: Academic Press, Inc.; 1984. p. 283-93.
- 12.Esterbauer H, Cheeseman KH, Determination of aldehydiclipid peroxidation products: malonaldehyde and 4-hydroxynonenal., In: Packer L and Glazer AN. Oxygen radicals in biological systemsSan Diego: Academic Press, Inc.; 1990. p. 407-21.
- 13.Montine TJ, Picklo MJ, Amarnath V, Whetsell Jr. WO, Graham DG. Neurotoxicity of endogenous cysteinylcatechols. Exp. Neurol. 1997; 148: 26-33.
- 14.Gluck MR, Zeevalk GD. Inhibition of brain mitochondrial respiration by dopamine and its metabolites: implications for Parkinson's disease and catecholamine-associated diseases. J. Neurochem. 2004; 91: 788-95.
- 15.Hodnick WF, Duval DL, Pardini RS. Inhibition of mitochondrial respiration and cyanide-stimulated generation of reactive oxygen species by selected flavonoids. Biochem. Pharmacol. 1994; 47: 573-80.
- 16.Nissinen E, Kaheinen P, Penttilä KE, Kaivola J, Lindén I-B. Entacapone, a novel catechol-O-methyltransferase inhibitor for Parkinson's disease, does not impair mitochondrial energy production. Eur. J. Pharmacol. 1997; 340: 287-94.
- 17.Korlipara LVP, Cooper JM, Schapira AHV. Diferences in toxicity of the catechol-O-methyl transferase inhibitors, tolcapone and entacapone to cultured human neuroblastoma cells. Neuropharmacology 2004; 46: 562-9.
- 18.Boveris A, Cadenas E, Stoppani AOM. Role of ubiquinone in the mitochondrial generation. Biochem J 1976; 156: 435-44.
- 19.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford: Oxford University Press.; 1998
- 20.Nutter LM, Wu Y-Y, Ngo EO, Sierra EE, Gutierrez PL, Abul-Hajj YJ. An o-quinone form of estrogen produces free radicals in human brest cancer cells: correlation with DNA damage. Chem. Res. Toxicol. 1994; 7: 23-8.
- 21.Jacobson JB, Duchen MR. MItochondrial oxidative stress and cell death in astrocytes - requirement for stored Ca2+ and sustained opening of the permeability transition pore. J. Cell Sci. 2002; 115: 1175-88.
- 22.Schweigert N, Zehnder AJB, Eggen RIL. Chemical properties of catechols and their molecules modes of toxic action in cells, from microorganisms to mammals. Environmental Microbiology 2001; 3: 81-91.
- 23.Koss G, Tesseraux I. Hydrocarbons, In: Marquardt SG, Schafer RM and Welsh F. ToxicologyCalifornia: Academic Press; 1999. p. 603-09.
- 24.Snyder R. Hedli CC. An overview of benzene metabolism. Environ Health Perspect 1996; 04 (Suppl 6): 116571.
- 25.Cotrim HP, de Freitas LAR, Freitas C, Braga L, Sousa R, Carvalho F, Paraná R, Santos-Jesus R, Andrade Z. Clinical and histopathological features of NASH in workers exposed to chemicals with or without associated metabolic conditions. Liver Int 2004; 24: 131-5.
Publication Dates
-
Publication in this collection
28 May 2007 -
Date of issue
2005